{"title":"Exploring the low-index surfaces of D52-La2O3 from the first-principles calculations","authors":"H.F. Sun, S.P. Sun, Y.R. Wang, Y. Zhang","doi":"10.1016/j.chemphys.2024.112418","DOIUrl":null,"url":null,"abstract":"<div><p>The surface relaxations, surface stability, electronic structures, and equilibrium morphology of D5<sub>2</sub>-La<sub>2</sub>O<sub>3</sub> were analyzed by means of first-principles calculations. The stoichiometric surfaces of D5<sub>2</sub>-La<sub>2</sub>O<sub>3</sub> possess thermodynamic energies of the following order: (0<!--> <!-->0<!--> <!-->1) < (1<!--> <!-->1<!--> <!-->0) < (1<!--> <!-->0<!--> <!-->0). Changes in temperature and the partial pressure of oxygen were employed to determine the energy of the non-stoichiometric surfaces. The results indicated that the energies of the (ns-1La1O)-terminated (1<!--> <!-->0<!--> <!-->0) and (ns-1La)-terminated (0<!--> <!-->0<!--> <!-->1) surfaces increased with increasing oxygen partial pressures and decreased with temperatures, whereas the (ns-1O)-terminated (0<!--> <!-->0<!--> <!-->1) and (ns-1O)-terminated (1<!--> <!-->0<!--> <!-->0) surfaces exhibited the reverse rule. According to the calculated density of states, surface relaxations primarily impact the surface electronic structures. The Gibbs-Wulff model was used to forecast the equilibrium morphology of D5<sub>2</sub>-La<sub>2</sub>O<sub>3</sub>, which followed in comparison with other’s experimental findings.</p></div>","PeriodicalId":272,"journal":{"name":"Chemical Physics","volume":"587 ","pages":"Article 112418"},"PeriodicalIF":2.0000,"publicationDate":"2024-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0301010424002477","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
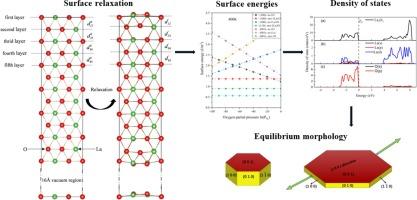
Abstract
The surface relaxations, surface stability, electronic structures, and equilibrium morphology of D52-La2O3 were analyzed by means of first-principles calculations. The stoichiometric surfaces of D52-La2O3 possess thermodynamic energies of the following order: (0 0 1) < (1 1 0) < (1 0 0). Changes in temperature and the partial pressure of oxygen were employed to determine the energy of the non-stoichiometric surfaces. The results indicated that the energies of the (ns-1La1O)-terminated (1 0 0) and (ns-1La)-terminated (0 0 1) surfaces increased with increasing oxygen partial pressures and decreased with temperatures, whereas the (ns-1O)-terminated (0 0 1) and (ns-1O)-terminated (1 0 0) surfaces exhibited the reverse rule. According to the calculated density of states, surface relaxations primarily impact the surface electronic structures. The Gibbs-Wulff model was used to forecast the equilibrium morphology of D52-La2O3, which followed in comparison with other’s experimental findings.
期刊介绍:
Chemical Physics publishes experimental and theoretical papers on all aspects of chemical physics. In this journal, experiments are related to theory, and in turn theoretical papers are related to present or future experiments. Subjects covered include: spectroscopy and molecular structure, interacting systems, relaxation phenomena, biological systems, materials, fundamental problems in molecular reactivity, molecular quantum theory and statistical mechanics. Computational chemistry studies of routine character are not appropriate for this journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
