Elucidating Adsorption Mechanisms and Characteristics of Emerging Aromatic Organic Contaminants to Graphene Material by Quantum Chemical Calculation Integrated with Interpretable Machine Learning
Thilini Maheshika Herath, Bei Zhang, Dhimas Dwinandha and Manabu Fujii*,
{"title":"Elucidating Adsorption Mechanisms and Characteristics of Emerging Aromatic Organic Contaminants to Graphene Material by Quantum Chemical Calculation Integrated with Interpretable Machine Learning","authors":"Thilini Maheshika Herath, Bei Zhang, Dhimas Dwinandha and Manabu Fujii*, ","doi":"10.1021/acsestwater.4c0021910.1021/acsestwater.4c00219","DOIUrl":null,"url":null,"abstract":"<p >As a complementary or alternative approach to experiments, theoretical computation of adsorption between carbon materials and emerging aromatic organic contaminants (AOCs) is increasingly important in elucidating adsorption mechanisms and characteristics, as well as their predictions. In this study, the adsorption energies between graphene and 112 AOCs were first analyzed by density functional theory (DFT-D). By the use of quantum molecular descriptors, different machine learning (ML) algorithms were developed. EXtreme gradient boosting exhibited the best performance among the four ML algorithms investigated, showing the lowest root-mean-square percentage error of 4.5% for the test data set. Accordingly, the interpretable ML technique (i.e., SHAP) assessed the importance and dependence of descriptors in the adsorption mechanisms of AOCs to graphene. The global interpretation confirmed that the molecular-volume-induced van der Waals interactions including π–π stacking are dominant, whereas the other interactions (e.g., induced hydrogen and electrostatic interactions) are comparably less significant in the adsorption of most AOCs on graphene. In contrast, using local interpretation, hydrogen bonds and induced dipole interactions with surrounding water were identified as important explanatory variables in the adsorption of AOCs containing carbonyl and sulfur functional groups. Therefore, the developed DFT-D-based ML models could be a reference model for theoretical and experimental studies.</p>","PeriodicalId":93847,"journal":{"name":"ACS ES&T water","volume":"4 9","pages":"3918–3930 3918–3930"},"PeriodicalIF":4.8000,"publicationDate":"2024-08-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS ES&T water","FirstCategoryId":"1085","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsestwater.4c00219","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENVIRONMENTAL SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
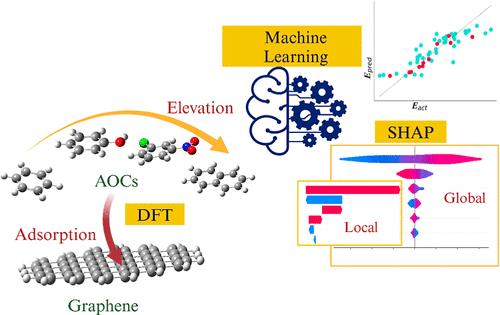
As a complementary or alternative approach to experiments, theoretical computation of adsorption between carbon materials and emerging aromatic organic contaminants (AOCs) is increasingly important in elucidating adsorption mechanisms and characteristics, as well as their predictions. In this study, the adsorption energies between graphene and 112 AOCs were first analyzed by density functional theory (DFT-D). By the use of quantum molecular descriptors, different machine learning (ML) algorithms were developed. EXtreme gradient boosting exhibited the best performance among the four ML algorithms investigated, showing the lowest root-mean-square percentage error of 4.5% for the test data set. Accordingly, the interpretable ML technique (i.e., SHAP) assessed the importance and dependence of descriptors in the adsorption mechanisms of AOCs to graphene. The global interpretation confirmed that the molecular-volume-induced van der Waals interactions including π–π stacking are dominant, whereas the other interactions (e.g., induced hydrogen and electrostatic interactions) are comparably less significant in the adsorption of most AOCs on graphene. In contrast, using local interpretation, hydrogen bonds and induced dipole interactions with surrounding water were identified as important explanatory variables in the adsorption of AOCs containing carbonyl and sulfur functional groups. Therefore, the developed DFT-D-based ML models could be a reference model for theoretical and experimental studies.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
