Fragment Addition-Based Design of Heteroaromatic-Biphenyl-DAPYs as Potent and Orally Available Non-nucleoside Reverse Transcriptase Inhibitors Featuring Significantly Enhanced Safety.
Wen-Juan Huang, Christophe Pannecouque, Erik De Clercq, Shuai Wang, Fen-Er Chen
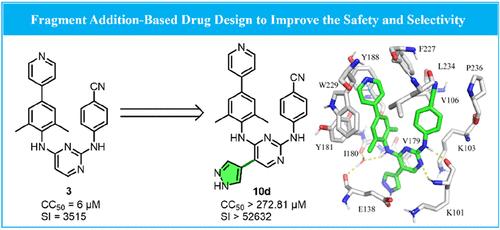
{"title":"Fragment Addition-Based Design of Heteroaromatic-Biphenyl-DAPYs as Potent and Orally Available Non-nucleoside Reverse Transcriptase Inhibitors Featuring Significantly Enhanced Safety.","authors":"Wen-Juan Huang, Christophe Pannecouque, Erik De Clercq, Shuai Wang, Fen-Er Chen","doi":"10.1021/acs.jmedchem.4c01571","DOIUrl":null,"url":null,"abstract":"<p><p>Our previously disclosed biphenyl-DAPY <b>3</b> emerged as a potent inhibitor against WT HIV-1 and various mutant strains. Yet, its journey toward clinical application was thwarted by pronounced cytotoxicity and low selectivity (CC<sub>50</sub> = 6 μM, SI = 3515). The safety improvement approach we employed in this work entailed the incorporation of diverse heteroaromatic substituents at the C5 position to exploit the tolerant regions of the NNRTIs' binding pocket through fragment addition-based drug design strategy, ultimately leading to the identification of a series of novel heteroaromatic-biphenyl-DAPYs. The exemplary compound <b>10d</b> revealed a striking reduction in cytotoxicity (CC<sub>50</sub> > 272.81 μM), nearly 45.5 times lower than <b>3</b>, while showcasing 15-fold increase in selectivity (SI > 52632). This analog sustained exceptional anti-HIV-1 activity against both WT HIV-1 (EC<sub>50</sub> = 5 nM) and various mutant strains. Compared to <b>3</b>, a markedly slower rate of metabolism in human liver microsomes of <b>10d</b> was observed. Its pharmacokinetic profile was equally captivating, featuring excellent oral bioavailability (<i>F</i> = 57.4%). Moreover, <b>10d</b> exhibited a delicate sensitivity toward CYP, minimal inhibition of hERG, and no detectable acute toxicity in vivo. These enchanting findings illuminated the potential of <b>10d</b> as a promising candidate for HIV-1 therapy.</p>","PeriodicalId":46,"journal":{"name":"Journal of Medicinal Chemistry","volume":" ","pages":"17568-17584"},"PeriodicalIF":6.8000,"publicationDate":"2024-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1021/acs.jmedchem.4c01571","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
Our previously disclosed biphenyl-DAPY 3 emerged as a potent inhibitor against WT HIV-1 and various mutant strains. Yet, its journey toward clinical application was thwarted by pronounced cytotoxicity and low selectivity (CC50 = 6 μM, SI = 3515). The safety improvement approach we employed in this work entailed the incorporation of diverse heteroaromatic substituents at the C5 position to exploit the tolerant regions of the NNRTIs' binding pocket through fragment addition-based drug design strategy, ultimately leading to the identification of a series of novel heteroaromatic-biphenyl-DAPYs. The exemplary compound 10d revealed a striking reduction in cytotoxicity (CC50 > 272.81 μM), nearly 45.5 times lower than 3, while showcasing 15-fold increase in selectivity (SI > 52632). This analog sustained exceptional anti-HIV-1 activity against both WT HIV-1 (EC50 = 5 nM) and various mutant strains. Compared to 3, a markedly slower rate of metabolism in human liver microsomes of 10d was observed. Its pharmacokinetic profile was equally captivating, featuring excellent oral bioavailability (F = 57.4%). Moreover, 10d exhibited a delicate sensitivity toward CYP, minimal inhibition of hERG, and no detectable acute toxicity in vivo. These enchanting findings illuminated the potential of 10d as a promising candidate for HIV-1 therapy.
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
