Rachael G Aubin, Javier Montelongo, Robert Hu, Elijah Gunther, Patrick Nicodemus, Pablo G Camara
{"title":"使用单细胞RNA-seq数据对大块组织中细胞丰度进行聚类独立估计。","authors":"Rachael G Aubin, Javier Montelongo, Robert Hu, Elijah Gunther, Patrick Nicodemus, Pablo G Camara","doi":"10.1101/2023.02.06.527318","DOIUrl":null,"url":null,"abstract":"<p><p>Single-cell RNA-sequencing has transformed the study of biological tissues by enabling transcriptomic characterizations of their constituent cell states. Computational methods for gene expression deconvolution use this information to infer the cell composition of related tissues profiled at the bulk level. However, current deconvolution methods are restricted to discrete cell types and have limited power to make inferences about continuous cellular processes like cell differentiation or immune cell activation. We present ConDecon, a clustering-independent method for inferring the likelihood for each cell in a single-cell dataset to be present in a bulk tissue. ConDecon represents an improvement in phenotypic resolution and functionality with respect to regression-based methods. Using ConDecon, we discover the implication of neurodegenerative microglia inflammatory pathways in the mesenchymal transformation of pediatric ependymoma and characterize their spatial trajectories of activation. The generality of this approach enables the deconvolution of other data modalities such as bulk ATAC-seq data.</p>","PeriodicalId":72407,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9934539/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clustering-independent estimation of cell abundances in bulk tissues using single-cell RNA-seq data.\",\"authors\":\"Rachael G Aubin, Javier Montelongo, Robert Hu, Elijah Gunther, Patrick Nicodemus, Pablo G Camara\",\"doi\":\"10.1101/2023.02.06.527318\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Single-cell RNA-sequencing has transformed the study of biological tissues by enabling transcriptomic characterizations of their constituent cell states. Computational methods for gene expression deconvolution use this information to infer the cell composition of related tissues profiled at the bulk level. However, current deconvolution methods are restricted to discrete cell types and have limited power to make inferences about continuous cellular processes like cell differentiation or immune cell activation. We present ConDecon, a clustering-independent method for inferring the likelihood for each cell in a single-cell dataset to be present in a bulk tissue. ConDecon represents an improvement in phenotypic resolution and functionality with respect to regression-based methods. Using ConDecon, we discover the implication of neurodegenerative microglia inflammatory pathways in the mesenchymal transformation of pediatric ependymoma and characterize their spatial trajectories of activation. The generality of this approach enables the deconvolution of other data modalities such as bulk ATAC-seq data.</p>\",\"PeriodicalId\":72407,\"journal\":{\"name\":\"bioRxiv : the preprint server for biology\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-09-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9934539/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"bioRxiv : the preprint server for biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/2023.02.06.527318\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.02.06.527318","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Clustering-independent estimation of cell abundances in bulk tissues using single-cell RNA-seq data.
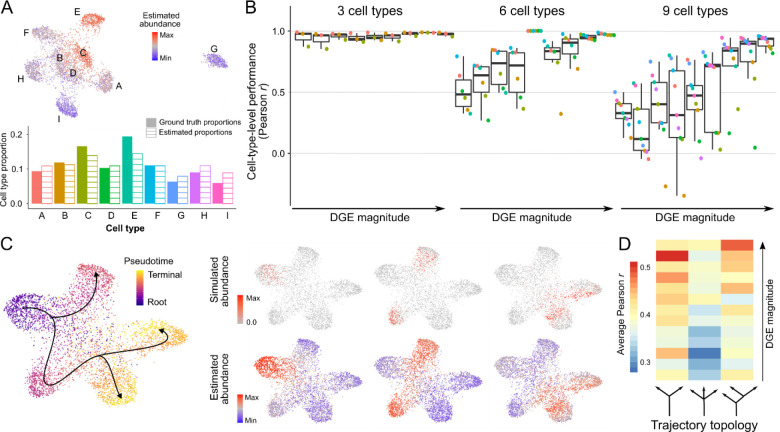
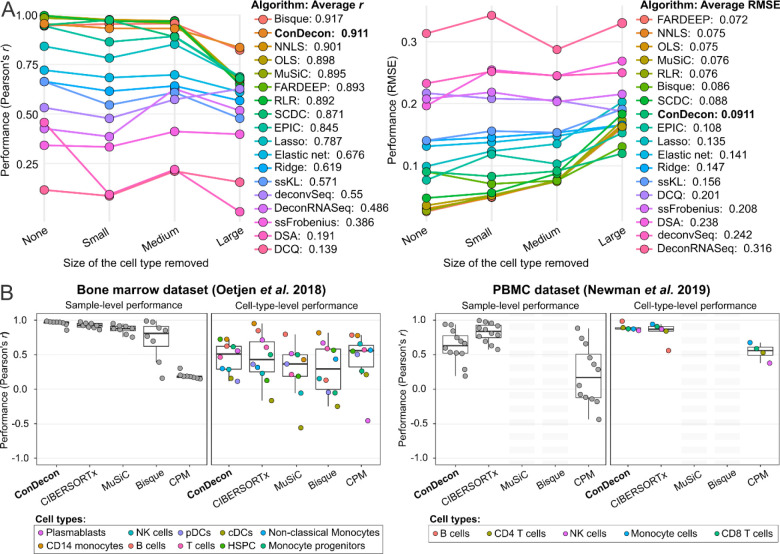
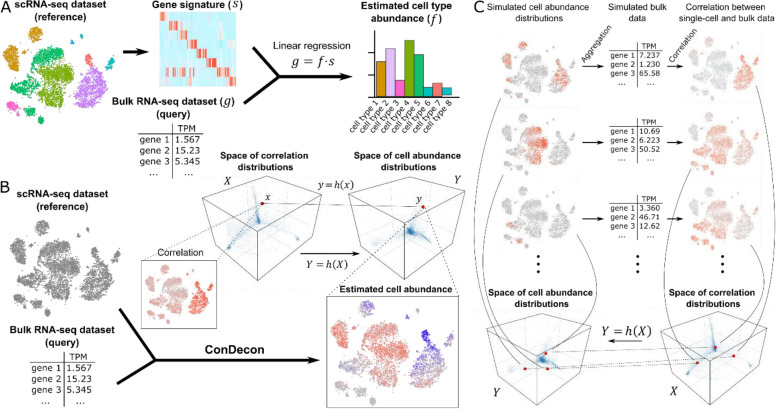
Single-cell RNA-sequencing has transformed the study of biological tissues by enabling transcriptomic characterizations of their constituent cell states. Computational methods for gene expression deconvolution use this information to infer the cell composition of related tissues profiled at the bulk level. However, current deconvolution methods are restricted to discrete cell types and have limited power to make inferences about continuous cellular processes like cell differentiation or immune cell activation. We present ConDecon, a clustering-independent method for inferring the likelihood for each cell in a single-cell dataset to be present in a bulk tissue. ConDecon represents an improvement in phenotypic resolution and functionality with respect to regression-based methods. Using ConDecon, we discover the implication of neurodegenerative microglia inflammatory pathways in the mesenchymal transformation of pediatric ependymoma and characterize their spatial trajectories of activation. The generality of this approach enables the deconvolution of other data modalities such as bulk ATAC-seq data.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
