{"title":"二氢嘧啶酶缺乏症患者的诊断奥德赛:病例报告和文献综述。","authors":"Daniah Albokhari, Ohood Alharbi, Alyssa Blesson, Mahim Jain","doi":"10.1101/mcs.a006319","DOIUrl":null,"url":null,"abstract":"<p><p>Dihydropyrimidinase (DHP) deficiency is an autosomal recessive metabolic disorder caused by biallelic pathogenic variants of <i>DPYS</i> Patients with DHP deficiency exhibit a broad spectrum of phenotypes, ranging from severe neurological and gastrointestinal involvement to cases with no apparent symptoms. The biochemical diagnosis of DHP deficiency is based on the detection of a significant amount of dihydropyrimidines in urine, plasma, and cerebrospinal fluid samples. Molecular genetic testing, specifically the identification of biallelic pathogenic variants in <i>DPYS</i>, has proven instrumental in confirming the diagnosis and facilitating family studies. This case study documents the diagnostic journey of an 18-yr-old patient with DHP deficiency, highlighting features at the severe end of the clinical spectrum. Notably, our patient exhibited previously unreported skeletal features that positively responded to bisphosphonate treatment, contributing valuable insights to the clinical characterization of DHP deficiency. Additionally, a novel <i>DPYS</i> variant was identified and confirmed pathogenicity through metabolic testing, further expanding the variant spectrum of the gene. Our case emphasizes the importance of a comprehensive diagnostic approach using genetic sequencing and metabolic testing for accurate diagnosis.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":"9 4","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10815279/pdf/","citationCount":"0","resultStr":"{\"title\":\"The diagnostic odyssey of a patient with dihydropyrimidinase deficiency: a case report and review of the literature.\",\"authors\":\"Daniah Albokhari, Ohood Alharbi, Alyssa Blesson, Mahim Jain\",\"doi\":\"10.1101/mcs.a006319\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Dihydropyrimidinase (DHP) deficiency is an autosomal recessive metabolic disorder caused by biallelic pathogenic variants of <i>DPYS</i> Patients with DHP deficiency exhibit a broad spectrum of phenotypes, ranging from severe neurological and gastrointestinal involvement to cases with no apparent symptoms. The biochemical diagnosis of DHP deficiency is based on the detection of a significant amount of dihydropyrimidines in urine, plasma, and cerebrospinal fluid samples. Molecular genetic testing, specifically the identification of biallelic pathogenic variants in <i>DPYS</i>, has proven instrumental in confirming the diagnosis and facilitating family studies. This case study documents the diagnostic journey of an 18-yr-old patient with DHP deficiency, highlighting features at the severe end of the clinical spectrum. Notably, our patient exhibited previously unreported skeletal features that positively responded to bisphosphonate treatment, contributing valuable insights to the clinical characterization of DHP deficiency. Additionally, a novel <i>DPYS</i> variant was identified and confirmed pathogenicity through metabolic testing, further expanding the variant spectrum of the gene. Our case emphasizes the importance of a comprehensive diagnostic approach using genetic sequencing and metabolic testing for accurate diagnosis.</p>\",\"PeriodicalId\":10360,\"journal\":{\"name\":\"Cold Spring Harbor Molecular Case Studies\",\"volume\":\"9 4\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-01-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10815279/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cold Spring Harbor Molecular Case Studies\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/mcs.a006319\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/12/1 0:00:00\",\"PubModel\":\"Print\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006319","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/1 0:00:00","PubModel":"Print","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
The diagnostic odyssey of a patient with dihydropyrimidinase deficiency: a case report and review of the literature.
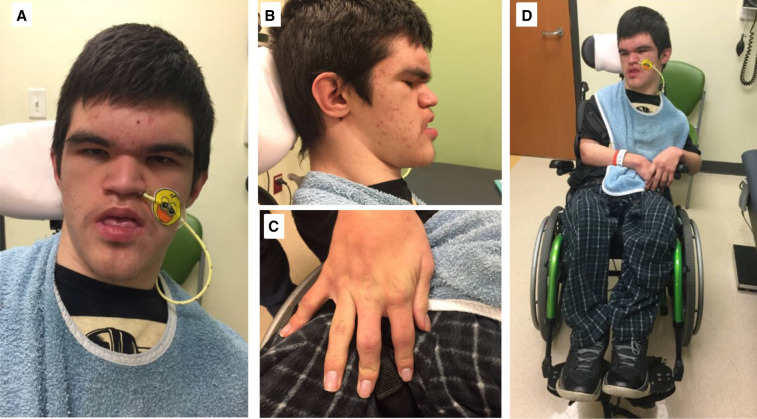
Dihydropyrimidinase (DHP) deficiency is an autosomal recessive metabolic disorder caused by biallelic pathogenic variants of DPYS Patients with DHP deficiency exhibit a broad spectrum of phenotypes, ranging from severe neurological and gastrointestinal involvement to cases with no apparent symptoms. The biochemical diagnosis of DHP deficiency is based on the detection of a significant amount of dihydropyrimidines in urine, plasma, and cerebrospinal fluid samples. Molecular genetic testing, specifically the identification of biallelic pathogenic variants in DPYS, has proven instrumental in confirming the diagnosis and facilitating family studies. This case study documents the diagnostic journey of an 18-yr-old patient with DHP deficiency, highlighting features at the severe end of the clinical spectrum. Notably, our patient exhibited previously unreported skeletal features that positively responded to bisphosphonate treatment, contributing valuable insights to the clinical characterization of DHP deficiency. Additionally, a novel DPYS variant was identified and confirmed pathogenicity through metabolic testing, further expanding the variant spectrum of the gene. Our case emphasizes the importance of a comprehensive diagnostic approach using genetic sequencing and metabolic testing for accurate diagnosis.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
