Changwei Liu, Congtao Wen, Zekai Zhang, Yuxin Chen, Huachao Yang, Jia-hui Li, Cheng Lian and Honglai Liu
{"title":"镍(111)表面的吸附变化:含氧官能团的电子密度多样性","authors":"Changwei Liu, Congtao Wen, Zekai Zhang, Yuxin Chen, Huachao Yang, Jia-hui Li, Cheng Lian and Honglai Liu","doi":"10.1039/D3ME00168G","DOIUrl":null,"url":null,"abstract":"<p >In the catalytic hydrodeoxygenation (HDO) upgrading process of biomass pyrolysis, adsorption behavior plays a crucial role in subsequent reaction processes. A comprehensive understanding of the interfacial behavior is essential for advancing novel biomaterials and commercial bio-oil. We initially establish their precise orientation on the Ni(111) surface and identify the preferential binding site by calculating the binding energy of eight model components (<em>n</em>-butanol, acetic acid, methyl acetate, <em>n</em>-hexanal, toluene, catechol, guaiacol, and 3-methyl-1,2-cyclopentanone). Differences in the electrostatic potential of functional groups and their interactions with the surface lead to surface electrostatic potential distributions, with compounds containing aldehyde functionality demonstrating increased reactivity. To account for competitive adsorption behavior among multiple molecules, ReaxFF-MD simulations were conducted to investigate the adsorption of guaiacol molecules. The inclusion of acetic acid enhances the polarization effect and non-uniformity, indicating competitive adsorption between guaiacol and acetic acid molecules. The chair conformation of acetic acid was demonstrated to be more reasonable from a kinetic perspective, leading to a stronger surface charge induction effect compared to guaiacol. Additionally, this non-uniform distribution is closely correlated with the characteristic bond activations of adsorbed active molecules, serving as a driving force to enhance further hydrogenation and deoxygenation activities of the molecules.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 5","pages":" 477-489"},"PeriodicalIF":3.2000,"publicationDate":"2024-02-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Adsorption variations on the Ni(111) surface: electron density diversity from oxygen-containing functional groups†\",\"authors\":\"Changwei Liu, Congtao Wen, Zekai Zhang, Yuxin Chen, Huachao Yang, Jia-hui Li, Cheng Lian and Honglai Liu\",\"doi\":\"10.1039/D3ME00168G\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In the catalytic hydrodeoxygenation (HDO) upgrading process of biomass pyrolysis, adsorption behavior plays a crucial role in subsequent reaction processes. A comprehensive understanding of the interfacial behavior is essential for advancing novel biomaterials and commercial bio-oil. We initially establish their precise orientation on the Ni(111) surface and identify the preferential binding site by calculating the binding energy of eight model components (<em>n</em>-butanol, acetic acid, methyl acetate, <em>n</em>-hexanal, toluene, catechol, guaiacol, and 3-methyl-1,2-cyclopentanone). Differences in the electrostatic potential of functional groups and their interactions with the surface lead to surface electrostatic potential distributions, with compounds containing aldehyde functionality demonstrating increased reactivity. To account for competitive adsorption behavior among multiple molecules, ReaxFF-MD simulations were conducted to investigate the adsorption of guaiacol molecules. The inclusion of acetic acid enhances the polarization effect and non-uniformity, indicating competitive adsorption between guaiacol and acetic acid molecules. The chair conformation of acetic acid was demonstrated to be more reasonable from a kinetic perspective, leading to a stronger surface charge induction effect compared to guaiacol. Additionally, this non-uniform distribution is closely correlated with the characteristic bond activations of adsorbed active molecules, serving as a driving force to enhance further hydrogenation and deoxygenation activities of the molecules.</p>\",\"PeriodicalId\":91,\"journal\":{\"name\":\"Molecular Systems Design & Engineering\",\"volume\":\" 5\",\"pages\":\" 477-489\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-02-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Systems Design & Engineering\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/me/d3me00168g\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/me/d3me00168g","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
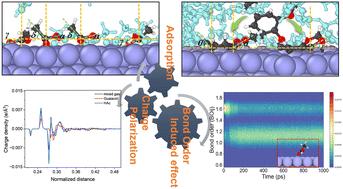
在生物质热解的催化加氢脱氧(HDO)升级过程中,吸附行为在后续反应过程中起着至关重要的作用。全面了解界面行为对于开发新型生物材料和商业生物油至关重要。我们通过计算八种模型成分(正丁醇、乙酸、乙酸甲酯、正己醛、甲苯、邻苯二酚、愈创木酚、3-甲基-1,2-环戊酮)的结合能,初步确定了它们在 Ni (111) 表面的精确取向,并确定了优先结合位点。官能团的静电电位及其与表面的相互作用差异导致了表面静电电位分布,含有醛官能团的化合物显示出更高的反应活性。为了解释多个分子之间的竞争吸附行为,我们进行了 ReaxFF-MD 模拟,以研究愈创木酚分子的吸附情况。醋酸的加入增强了极化效应和不均匀性,表明愈创木酚和醋酸分子之间存在竞争性吸附。从动力学角度看,乙酸的椅子构象更合理,因此与愈创木酚相比,其表面电荷诱导效应更强。此外,这种不均匀分布与吸附的活性分子的键活化特征密切相关,成为进一步提高分子氢化和脱氧活性的驱动力。
Adsorption variations on the Ni(111) surface: electron density diversity from oxygen-containing functional groups†
In the catalytic hydrodeoxygenation (HDO) upgrading process of biomass pyrolysis, adsorption behavior plays a crucial role in subsequent reaction processes. A comprehensive understanding of the interfacial behavior is essential for advancing novel biomaterials and commercial bio-oil. We initially establish their precise orientation on the Ni(111) surface and identify the preferential binding site by calculating the binding energy of eight model components (n-butanol, acetic acid, methyl acetate, n-hexanal, toluene, catechol, guaiacol, and 3-methyl-1,2-cyclopentanone). Differences in the electrostatic potential of functional groups and their interactions with the surface lead to surface electrostatic potential distributions, with compounds containing aldehyde functionality demonstrating increased reactivity. To account for competitive adsorption behavior among multiple molecules, ReaxFF-MD simulations were conducted to investigate the adsorption of guaiacol molecules. The inclusion of acetic acid enhances the polarization effect and non-uniformity, indicating competitive adsorption between guaiacol and acetic acid molecules. The chair conformation of acetic acid was demonstrated to be more reasonable from a kinetic perspective, leading to a stronger surface charge induction effect compared to guaiacol. Additionally, this non-uniform distribution is closely correlated with the characteristic bond activations of adsorbed active molecules, serving as a driving force to enhance further hydrogenation and deoxygenation activities of the molecules.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
