Line Aagaard Nolting, Tess Holling, Gen Nishimura, Jakob Ek, Mads Bak, Merete Ljungberg, Kerstin Kutsche, Hanne Hove
{"title":"新的双链PISD错义变体会导致脊柱软骨发育不良,并伴有不成比例的矮小身材和线粒体形态破碎。","authors":"Line Aagaard Nolting, Tess Holling, Gen Nishimura, Jakob Ek, Mads Bak, Merete Ljungberg, Kerstin Kutsche, Hanne Hove","doi":"10.1111/cge.14549","DOIUrl":null,"url":null,"abstract":"<p>Biallelic variants in <i>PISD</i> cause a phenotypic spectrum ranging from short stature with spondyloepimetaphyseal dysplasia (SEMD) to a multisystem disorder affecting eyes, ears, bones, and brain. <i>PISD</i> encodes the mitochondrial-localized enzyme phosphatidylserine decarboxylase. The PISD precursor is self-cleaved to generate a heteromeric mature enzyme that converts phosphatidylserine to the phospholipid phosphatidylethanolamine. We describe a 17-year-old male patient, born to unrelated healthy parents, with disproportionate short stature and SEMD, featuring platyspondyly, prominent epiphyses, and metaphyseal dysplasia. Trio genome sequencing revealed compound heterozygous <i>PISD</i> variants c.569C>T; p.(Ser190Leu) and c.799C>T; p.(His267Tyr) in the patient. Investigation of fibroblasts showed similar levels of the PISD precursor protein in both patient and control cells. However, patient cells had a significantly higher proportion of fragmented mitochondria compared to control cells cultured under basal condition and after treatment with 2-deoxyglucose that represses glycolysis and stimulates respiration. Structural data from the PISD orthologue in <i>Escherichia coli</i> suggest that the amino acid substitutions Ser190Leu and His267Tyr likely impair PISD's autoprocessing activity and/or phosphatidylethanolamine biosynthesis. Based on the data, we propose that the novel <i>PISD</i> p.(Ser190Leu) and p.(His267Tyr) variants likely act as hypomorphs and underlie the pure skeletal phenotype in the patient.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"106 3","pages":"360-366"},"PeriodicalIF":2.9000,"publicationDate":"2024-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14549","citationCount":"0","resultStr":"{\"title\":\"Novel biallelic PISD missense variants cause spondyloepimetaphyseal dysplasia with disproportionate short stature and fragmented mitochondrial morphology\",\"authors\":\"Line Aagaard Nolting, Tess Holling, Gen Nishimura, Jakob Ek, Mads Bak, Merete Ljungberg, Kerstin Kutsche, Hanne Hove\",\"doi\":\"10.1111/cge.14549\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Biallelic variants in <i>PISD</i> cause a phenotypic spectrum ranging from short stature with spondyloepimetaphyseal dysplasia (SEMD) to a multisystem disorder affecting eyes, ears, bones, and brain. <i>PISD</i> encodes the mitochondrial-localized enzyme phosphatidylserine decarboxylase. The PISD precursor is self-cleaved to generate a heteromeric mature enzyme that converts phosphatidylserine to the phospholipid phosphatidylethanolamine. We describe a 17-year-old male patient, born to unrelated healthy parents, with disproportionate short stature and SEMD, featuring platyspondyly, prominent epiphyses, and metaphyseal dysplasia. Trio genome sequencing revealed compound heterozygous <i>PISD</i> variants c.569C>T; p.(Ser190Leu) and c.799C>T; p.(His267Tyr) in the patient. Investigation of fibroblasts showed similar levels of the PISD precursor protein in both patient and control cells. However, patient cells had a significantly higher proportion of fragmented mitochondria compared to control cells cultured under basal condition and after treatment with 2-deoxyglucose that represses glycolysis and stimulates respiration. Structural data from the PISD orthologue in <i>Escherichia coli</i> suggest that the amino acid substitutions Ser190Leu and His267Tyr likely impair PISD's autoprocessing activity and/or phosphatidylethanolamine biosynthesis. Based on the data, we propose that the novel <i>PISD</i> p.(Ser190Leu) and p.(His267Tyr) variants likely act as hypomorphs and underlie the pure skeletal phenotype in the patient.</p>\",\"PeriodicalId\":10354,\"journal\":{\"name\":\"Clinical Genetics\",\"volume\":\"106 3\",\"pages\":\"360-366\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-05-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14549\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cge.14549\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14549","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Novel biallelic PISD missense variants cause spondyloepimetaphyseal dysplasia with disproportionate short stature and fragmented mitochondrial morphology
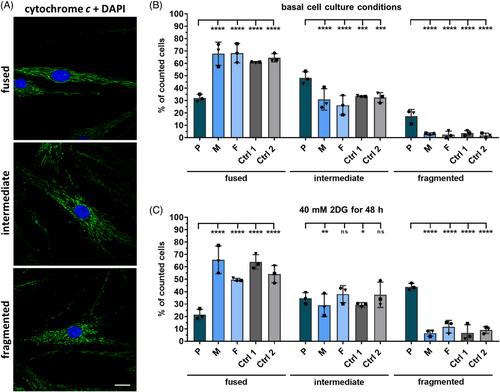
Biallelic variants in PISD cause a phenotypic spectrum ranging from short stature with spondyloepimetaphyseal dysplasia (SEMD) to a multisystem disorder affecting eyes, ears, bones, and brain. PISD encodes the mitochondrial-localized enzyme phosphatidylserine decarboxylase. The PISD precursor is self-cleaved to generate a heteromeric mature enzyme that converts phosphatidylserine to the phospholipid phosphatidylethanolamine. We describe a 17-year-old male patient, born to unrelated healthy parents, with disproportionate short stature and SEMD, featuring platyspondyly, prominent epiphyses, and metaphyseal dysplasia. Trio genome sequencing revealed compound heterozygous PISD variants c.569C>T; p.(Ser190Leu) and c.799C>T; p.(His267Tyr) in the patient. Investigation of fibroblasts showed similar levels of the PISD precursor protein in both patient and control cells. However, patient cells had a significantly higher proportion of fragmented mitochondria compared to control cells cultured under basal condition and after treatment with 2-deoxyglucose that represses glycolysis and stimulates respiration. Structural data from the PISD orthologue in Escherichia coli suggest that the amino acid substitutions Ser190Leu and His267Tyr likely impair PISD's autoprocessing activity and/or phosphatidylethanolamine biosynthesis. Based on the data, we propose that the novel PISD p.(Ser190Leu) and p.(His267Tyr) variants likely act as hypomorphs and underlie the pure skeletal phenotype in the patient.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
