Johanna P. Carbone, Andreas Irmler, Alejandro Gallo, Tobias Schäfer, William Z. Van Benschoten, James J. Shepherd and Andreas Grüneis
{"title":"利用周期耦合团簇理论研究 CO 在 Pt(111) 上的吸附。","authors":"Johanna P. Carbone, Andreas Irmler, Alejandro Gallo, Tobias Schäfer, William Z. Van Benschoten, James J. Shepherd and Andreas Grüneis","doi":"10.1039/D4FD00085D","DOIUrl":null,"url":null,"abstract":"<p >We present an application of periodic coupled-cluster theory to the calculation of CO adsorption energies on the Pt(111) surface for different adsorption sites. The calculations employ a range of recently developed theoretical and computational methods. In particular, we use a recently introduced coupled-cluster ansatz, denoted as CCSD(cT), to compute correlation energies of the metallic Pt surface with and without adsorbed CO molecules. The convergence of Hartree–Fock adsorption energy contributions with respect to randomly shifted <em>k</em>-meshes is discussed. Recently introduced basis set incompleteness error corrections make it possible to achieve well-converged correlation energy contributions to the adsorption energies. We show that CCSD(cT) theory predicts the correct order of adsorption energies for the considered adsorption sites. Furthermore, we find that binding of the CO molecule to the top and fcc site is dominated by Hartree–Fock and correlation energy contributions, respectively.</p>","PeriodicalId":49075,"journal":{"name":"Faraday Discussions","volume":"254 ","pages":" 586-597"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11339635/pdf/","citationCount":"0","resultStr":"{\"title\":\"CO adsorption on Pt(111) studied by periodic coupled cluster theory\",\"authors\":\"Johanna P. Carbone, Andreas Irmler, Alejandro Gallo, Tobias Schäfer, William Z. Van Benschoten, James J. Shepherd and Andreas Grüneis\",\"doi\":\"10.1039/D4FD00085D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We present an application of periodic coupled-cluster theory to the calculation of CO adsorption energies on the Pt(111) surface for different adsorption sites. The calculations employ a range of recently developed theoretical and computational methods. In particular, we use a recently introduced coupled-cluster ansatz, denoted as CCSD(cT), to compute correlation energies of the metallic Pt surface with and without adsorbed CO molecules. The convergence of Hartree–Fock adsorption energy contributions with respect to randomly shifted <em>k</em>-meshes is discussed. Recently introduced basis set incompleteness error corrections make it possible to achieve well-converged correlation energy contributions to the adsorption energies. We show that CCSD(cT) theory predicts the correct order of adsorption energies for the considered adsorption sites. Furthermore, we find that binding of the CO molecule to the top and fcc site is dominated by Hartree–Fock and correlation energy contributions, respectively.</p>\",\"PeriodicalId\":49075,\"journal\":{\"name\":\"Faraday Discussions\",\"volume\":\"254 \",\"pages\":\" 586-597\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-08-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11339635/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Faraday Discussions\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00085d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Faraday Discussions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00085d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 0
摘要
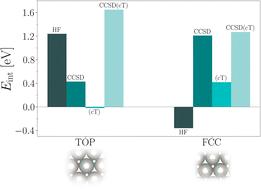
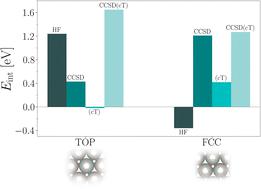
我们介绍了周期耦合簇理论在 Pt(111) 表面不同吸附位点 CO 吸附能计算中的应用。计算采用了一系列最新开发的理论和计算方法。特别是,我们使用了最近引入的耦合簇变量(CCSD(cT))来计算金属铂表面吸附和不吸附 CO 分子时的相关能。讨论了哈特里-福克吸附能贡献与随机偏移 k 型的收敛性。最近引入的基集不完备性误差修正使吸附能的相关能贡献达到良好的收敛性成为可能。我们表明,CCSD(cT)理论预测了所考虑的吸附位点的吸附能的正确顺序。此外,我们还发现 CO 分子与顶部和 fcc 位点的结合分别由 Hartree-Fock 和相关能贡献所主导。
CO adsorption on Pt(111) studied by periodic coupled cluster theory
We present an application of periodic coupled-cluster theory to the calculation of CO adsorption energies on the Pt(111) surface for different adsorption sites. The calculations employ a range of recently developed theoretical and computational methods. In particular, we use a recently introduced coupled-cluster ansatz, denoted as CCSD(cT), to compute correlation energies of the metallic Pt surface with and without adsorbed CO molecules. The convergence of Hartree–Fock adsorption energy contributions with respect to randomly shifted k-meshes is discussed. Recently introduced basis set incompleteness error corrections make it possible to achieve well-converged correlation energy contributions to the adsorption energies. We show that CCSD(cT) theory predicts the correct order of adsorption energies for the considered adsorption sites. Furthermore, we find that binding of the CO molecule to the top and fcc site is dominated by Hartree–Fock and correlation energy contributions, respectively.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
