Mariam Goubran, Ayodeji Aderibigbe, Emmanuel Jacquemin, Catherine Guettier, Safwat Girgis, Vincent Bain, Andrew L Mason
{"title":"病例报告:肝移植 15 年后诊断出带有复合杂合子 ABCB4 变体的进行性家族性肝内胆汁淤积 3 型。","authors":"Mariam Goubran, Ayodeji Aderibigbe, Emmanuel Jacquemin, Catherine Guettier, Safwat Girgis, Vincent Bain, Andrew L Mason","doi":"10.1186/s12881-020-01173-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Progressive familial intrahepatic cholestasis (PFIC) type 3 is an autosomal recessive disorder arising from mutations in the ATP-binding cassette subfamily B member 4 (ABCB4) gene. This gene encodes multidrug resistance protein-3 (MDR3) that acts as a hepatocanalicular floppase that transports phosphatidylcholine from the inner to the outer canalicular membrane. In the absence of phosphatidylcholine, the detergent activity of bile salts is amplified and this leads to cholangiopathy, bile duct loss and biliary cirrhosis. Patients usually present in infancy or childhood and often progress to end-stage liver disease before adulthood.</p><p><strong>Case presentation: </strong>We report a 32-year-old female who required cadaveric liver transplantation at the age of 17 for cryptogenic cirrhosis. When the patient developed chronic ductopenia in the allograft 15 years later, we hypothesized that the patient's original disease was due to a deficiency of a biliary transport protein and the ductopenia could be explained by an autoimmune response to neoantigen that was not previously encountered by the immune system. We therefore performed genetic analyses and immunohistochemistry of the native liver, which led to a diagnosis of PFIC3. However, there was no evidence of humoral immune response to the MDR3 and therefore, we assumed that the ductopenia observed in the allograft was likely due to chronic rejection rather than autoimmune disease in the allograft.</p><p><strong>Conclusions: </strong>Teenage patients referred for liver transplantation with cryptogenic liver disease should undergo work up for PFIC3. An accurate diagnosis of PFIC 3 is key for optimal management, therapeutic intervention, and avoidance of complications before the onset of end-stage liver disease.</p>","PeriodicalId":9015,"journal":{"name":"BMC Medical Genetics","volume":" ","pages":"238"},"PeriodicalIF":0.0000,"publicationDate":"2020-11-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7708126/pdf/","citationCount":"0","resultStr":"{\"title\":\"Case report: progressive familial intrahepatic cholestasis type 3 with compound heterozygous ABCB4 variants diagnosed 15 years after liver transplantation.\",\"authors\":\"Mariam Goubran, Ayodeji Aderibigbe, Emmanuel Jacquemin, Catherine Guettier, Safwat Girgis, Vincent Bain, Andrew L Mason\",\"doi\":\"10.1186/s12881-020-01173-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Progressive familial intrahepatic cholestasis (PFIC) type 3 is an autosomal recessive disorder arising from mutations in the ATP-binding cassette subfamily B member 4 (ABCB4) gene. This gene encodes multidrug resistance protein-3 (MDR3) that acts as a hepatocanalicular floppase that transports phosphatidylcholine from the inner to the outer canalicular membrane. In the absence of phosphatidylcholine, the detergent activity of bile salts is amplified and this leads to cholangiopathy, bile duct loss and biliary cirrhosis. Patients usually present in infancy or childhood and often progress to end-stage liver disease before adulthood.</p><p><strong>Case presentation: </strong>We report a 32-year-old female who required cadaveric liver transplantation at the age of 17 for cryptogenic cirrhosis. When the patient developed chronic ductopenia in the allograft 15 years later, we hypothesized that the patient's original disease was due to a deficiency of a biliary transport protein and the ductopenia could be explained by an autoimmune response to neoantigen that was not previously encountered by the immune system. We therefore performed genetic analyses and immunohistochemistry of the native liver, which led to a diagnosis of PFIC3. However, there was no evidence of humoral immune response to the MDR3 and therefore, we assumed that the ductopenia observed in the allograft was likely due to chronic rejection rather than autoimmune disease in the allograft.</p><p><strong>Conclusions: </strong>Teenage patients referred for liver transplantation with cryptogenic liver disease should undergo work up for PFIC3. An accurate diagnosis of PFIC 3 is key for optimal management, therapeutic intervention, and avoidance of complications before the onset of end-stage liver disease.</p>\",\"PeriodicalId\":9015,\"journal\":{\"name\":\"BMC Medical Genetics\",\"volume\":\" \",\"pages\":\"238\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-11-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7708126/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Medical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12881-020-01173-0\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12881-020-01173-0","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
摘要
背景:进行性家族性肝内胆汁淤积症(PFIC)3 型是一种常染色体隐性遗传疾病,由 ATP 结合盒 B 亚家族成员 4(ABCB4)基因突变引起。该基因编码多药耐药蛋白-3 (MDR3),它是一种肝管膜翻转酶,能将磷脂酰胆碱从肝管内膜转运到肝管外膜。在缺乏磷脂酰胆碱的情况下,胆盐的洗涤活性会被放大,从而导致胆管病变、胆管缺失和胆汁性肝硬化。患者通常在婴儿期或儿童期发病,通常在成年前发展为终末期肝病:我们报告了一名 32 岁的女性患者,她在 17 岁时因隐源性肝硬化而需要进行尸体肝移植。15 年后,患者在异体肝移植后出现慢性导管减少症,我们推测患者最初的疾病是由于胆汁转运蛋白缺乏所致,而导管减少症的原因可能是免疫系统对以前未曾接触过的新抗原产生了自身免疫反应。因此,我们对原生肝脏进行了基因分析和免疫组化,最终确诊为 PFIC3。然而,没有证据表明存在对MDR3的体液免疫反应,因此,我们认为在异体肝移植中观察到的导管减少症很可能是由于慢性排斥反应所致,而非异体肝移植中的自身免疫性疾病:结论:因隐源性肝病而转诊接受肝移植的青少年患者应进行PFIC3检查。在终末期肝病发生之前,准确诊断 PFIC3 是优化管理、治疗干预和避免并发症的关键。
Case report: progressive familial intrahepatic cholestasis type 3 with compound heterozygous ABCB4 variants diagnosed 15 years after liver transplantation.
Background: Progressive familial intrahepatic cholestasis (PFIC) type 3 is an autosomal recessive disorder arising from mutations in the ATP-binding cassette subfamily B member 4 (ABCB4) gene. This gene encodes multidrug resistance protein-3 (MDR3) that acts as a hepatocanalicular floppase that transports phosphatidylcholine from the inner to the outer canalicular membrane. In the absence of phosphatidylcholine, the detergent activity of bile salts is amplified and this leads to cholangiopathy, bile duct loss and biliary cirrhosis. Patients usually present in infancy or childhood and often progress to end-stage liver disease before adulthood.
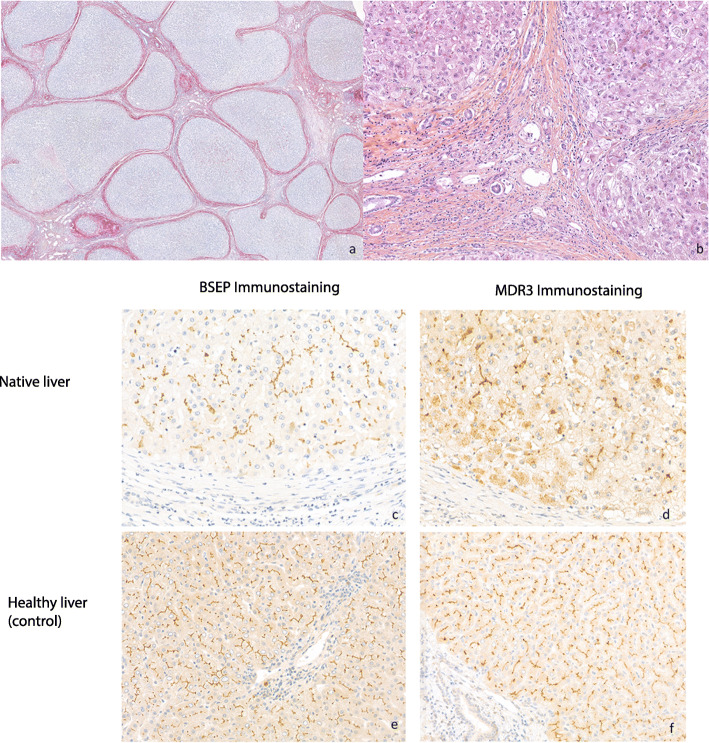
Case presentation: We report a 32-year-old female who required cadaveric liver transplantation at the age of 17 for cryptogenic cirrhosis. When the patient developed chronic ductopenia in the allograft 15 years later, we hypothesized that the patient's original disease was due to a deficiency of a biliary transport protein and the ductopenia could be explained by an autoimmune response to neoantigen that was not previously encountered by the immune system. We therefore performed genetic analyses and immunohistochemistry of the native liver, which led to a diagnosis of PFIC3. However, there was no evidence of humoral immune response to the MDR3 and therefore, we assumed that the ductopenia observed in the allograft was likely due to chronic rejection rather than autoimmune disease in the allograft.
Conclusions: Teenage patients referred for liver transplantation with cryptogenic liver disease should undergo work up for PFIC3. An accurate diagnosis of PFIC 3 is key for optimal management, therapeutic intervention, and avoidance of complications before the onset of end-stage liver disease.
期刊介绍:
BMC Medical Genetics is an open access journal publishing original peer-reviewed research articles in the effects of genetic variation in individuals, families and among populations in relation to human health and disease.
Note: BMC Medical Genetics is now closed. This journal has merged with BMC Medical Genomics, a broad-scope, open access community journal for all medical genetics and genomics research.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
