Joana Damásio, Ana Sardoeira, Maria Araújo, Isabel Carvalho, Jorge Sequeiros, José Barros
{"title":"弗里德赖希共济失调少见严重盲聋1例。","authors":"Joana Damásio, Ana Sardoeira, Maria Araújo, Isabel Carvalho, Jorge Sequeiros, José Barros","doi":"10.1186/s40673-021-00140-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Friedreich ataxia is the most frequent hereditary ataxia worldwide. Subclinical visual and auditory involvement has been recognized in these patients, with co-occurrence of severe blindness and deafness being rare.</p><p><strong>Case report: </strong>We describe a patient, homozygous for a 873 GAA expansion in the FXN gene, whose first symptoms appeared by the age of 8. At 22 years-old he developed sensorineural deafness, and at 26 visual impairment. Deafness had a progressive course over 11 years, until a stage of extreme severity which hindered communication. Visual acuity had a catastrophic deterioration, with blindness 3 years after visual impairment was first noticed. Audiograms documented progressive sensorineural deafness, most striking for low frequencies. Visual evoked potentials disclosed bilaterally increased P100 latency. He passed away at the age of 41 years old, at a stage of extreme disability, blind and deaf, in addition to the complete phenotype of a patient with Friedreich ataxia of more than 30 years duration.</p><p><strong>Discussion: </strong>Severe vision loss and extreme deafness has been described in very few patients with Friedreich ataxia. Long duration, severe disease and large expanded alleles may account for such an extreme phenotype; nonetheless, the role of factors as modifying genes warrants further investigation in this subset of patients.</p>","PeriodicalId":36752,"journal":{"name":"Cerebellum and Ataxias","volume":" ","pages":"17"},"PeriodicalIF":0.0000,"publicationDate":"2021-07-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40673-021-00140-6","citationCount":"3","resultStr":"{\"title\":\"Rare occurrence of severe blindness and deafness in Friedreich ataxia: a case report.\",\"authors\":\"Joana Damásio, Ana Sardoeira, Maria Araújo, Isabel Carvalho, Jorge Sequeiros, José Barros\",\"doi\":\"10.1186/s40673-021-00140-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Friedreich ataxia is the most frequent hereditary ataxia worldwide. Subclinical visual and auditory involvement has been recognized in these patients, with co-occurrence of severe blindness and deafness being rare.</p><p><strong>Case report: </strong>We describe a patient, homozygous for a 873 GAA expansion in the FXN gene, whose first symptoms appeared by the age of 8. At 22 years-old he developed sensorineural deafness, and at 26 visual impairment. Deafness had a progressive course over 11 years, until a stage of extreme severity which hindered communication. Visual acuity had a catastrophic deterioration, with blindness 3 years after visual impairment was first noticed. Audiograms documented progressive sensorineural deafness, most striking for low frequencies. Visual evoked potentials disclosed bilaterally increased P100 latency. He passed away at the age of 41 years old, at a stage of extreme disability, blind and deaf, in addition to the complete phenotype of a patient with Friedreich ataxia of more than 30 years duration.</p><p><strong>Discussion: </strong>Severe vision loss and extreme deafness has been described in very few patients with Friedreich ataxia. Long duration, severe disease and large expanded alleles may account for such an extreme phenotype; nonetheless, the role of factors as modifying genes warrants further investigation in this subset of patients.</p>\",\"PeriodicalId\":36752,\"journal\":{\"name\":\"Cerebellum and Ataxias\",\"volume\":\" \",\"pages\":\"17\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-07-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s40673-021-00140-6\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cerebellum and Ataxias\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s40673-021-00140-6\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cerebellum and Ataxias","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40673-021-00140-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
Rare occurrence of severe blindness and deafness in Friedreich ataxia: a case report.
Background: Friedreich ataxia is the most frequent hereditary ataxia worldwide. Subclinical visual and auditory involvement has been recognized in these patients, with co-occurrence of severe blindness and deafness being rare.
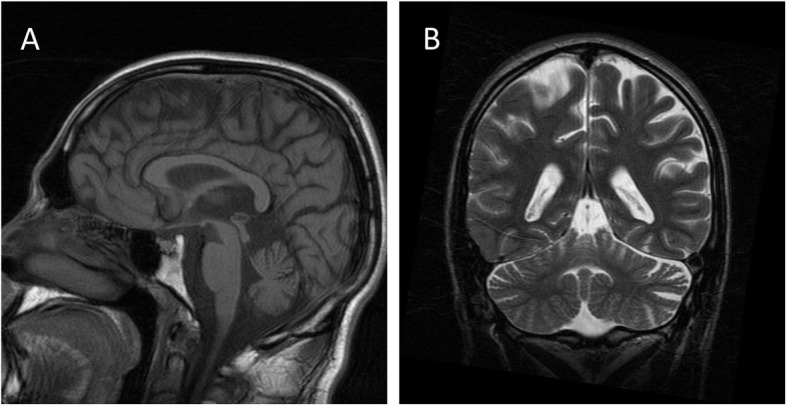
Case report: We describe a patient, homozygous for a 873 GAA expansion in the FXN gene, whose first symptoms appeared by the age of 8. At 22 years-old he developed sensorineural deafness, and at 26 visual impairment. Deafness had a progressive course over 11 years, until a stage of extreme severity which hindered communication. Visual acuity had a catastrophic deterioration, with blindness 3 years after visual impairment was first noticed. Audiograms documented progressive sensorineural deafness, most striking for low frequencies. Visual evoked potentials disclosed bilaterally increased P100 latency. He passed away at the age of 41 years old, at a stage of extreme disability, blind and deaf, in addition to the complete phenotype of a patient with Friedreich ataxia of more than 30 years duration.
Discussion: Severe vision loss and extreme deafness has been described in very few patients with Friedreich ataxia. Long duration, severe disease and large expanded alleles may account for such an extreme phenotype; nonetheless, the role of factors as modifying genes warrants further investigation in this subset of patients.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
