{"title":"利用UKBEC数据集进行表达微阵列与rna测序的比较,鉴定出一个与黑质MPZ基因相关的反式eqtl。","authors":"Letitia M F Sng, Peter C Thomson, Daniah Trabzuni","doi":"","DOIUrl":null,"url":null,"abstract":"<p><p>In recent years, the advantages of RNA-sequencing (RNA-Seq) have made it the platform of choice for measuring gene expression over traditional microarrays. However, RNA-Seq comes with bioinformatical challenges and higher computational costs. Therefore, this study set out to assess whether the increased depth of transcriptomic information facilitated by RNA-Seq is worth the increased computation over microarrays, specifically at three levels: absolute expression levels, differentially expressed genes identification, and expression QTL (eQTL) mapping in regions of the human brain. Using the United Kingdom Brain Expression Consortium (UKBEC) dataset, there is high agreement of gene expression levels measured by microarrays and RNA-seq when quantifying absolute expression levels and when identifying differentially expressed genes. These findings suggest that depending on the aims of a study, the relative ease of working with microarray data may outweigh the computational time and costs of RNA-Seq pipelines. On the other, there was low agreement when mapping eQTLs. However, a number of eQTLs associated with genes that play important roles in the brain were found in both platforms. For example, a <i>trans</i>-eQTL was mapped that is associated with the <i>MPZ</i> gene in the substantia nigra. These eQTLs that we have highlighted are extremely promising candidates that merit further investigation.</p>","PeriodicalId":93097,"journal":{"name":"Frontiers in neurology and neuroscience research","volume":"1 ","pages":"100001"},"PeriodicalIF":0.0000,"publicationDate":"2020-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7611373/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comparison Between Expression Microarrays and RNA-Sequencing Using UKBEC Dataset Identified a <i>trans</i>-eQTL Associated with <i>MPZ</i> Gene in Substantia Nigra.\",\"authors\":\"Letitia M F Sng, Peter C Thomson, Daniah Trabzuni\",\"doi\":\"\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>In recent years, the advantages of RNA-sequencing (RNA-Seq) have made it the platform of choice for measuring gene expression over traditional microarrays. However, RNA-Seq comes with bioinformatical challenges and higher computational costs. Therefore, this study set out to assess whether the increased depth of transcriptomic information facilitated by RNA-Seq is worth the increased computation over microarrays, specifically at three levels: absolute expression levels, differentially expressed genes identification, and expression QTL (eQTL) mapping in regions of the human brain. Using the United Kingdom Brain Expression Consortium (UKBEC) dataset, there is high agreement of gene expression levels measured by microarrays and RNA-seq when quantifying absolute expression levels and when identifying differentially expressed genes. These findings suggest that depending on the aims of a study, the relative ease of working with microarray data may outweigh the computational time and costs of RNA-Seq pipelines. On the other, there was low agreement when mapping eQTLs. However, a number of eQTLs associated with genes that play important roles in the brain were found in both platforms. For example, a <i>trans</i>-eQTL was mapped that is associated with the <i>MPZ</i> gene in the substantia nigra. These eQTLs that we have highlighted are extremely promising candidates that merit further investigation.</p>\",\"PeriodicalId\":93097,\"journal\":{\"name\":\"Frontiers in neurology and neuroscience research\",\"volume\":\"1 \",\"pages\":\"100001\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7611373/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in neurology and neuroscience research\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in neurology and neuroscience research","FirstCategoryId":"1085","ListUrlMain":"","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Comparison Between Expression Microarrays and RNA-Sequencing Using UKBEC Dataset Identified a trans-eQTL Associated with MPZ Gene in Substantia Nigra.
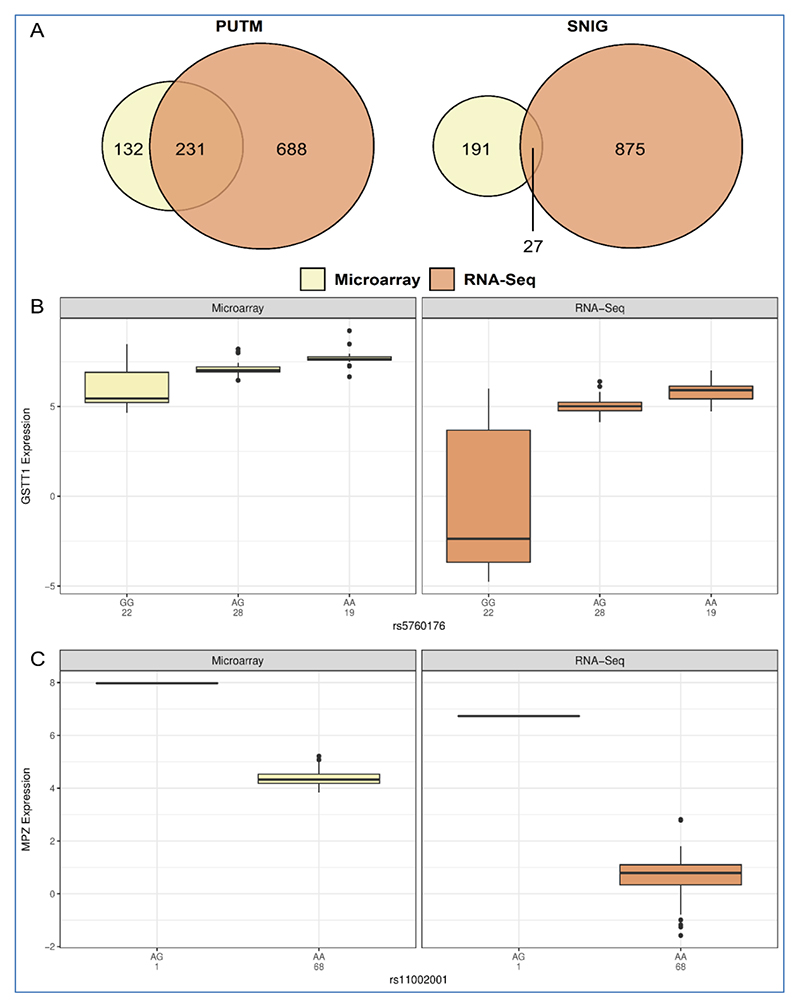
In recent years, the advantages of RNA-sequencing (RNA-Seq) have made it the platform of choice for measuring gene expression over traditional microarrays. However, RNA-Seq comes with bioinformatical challenges and higher computational costs. Therefore, this study set out to assess whether the increased depth of transcriptomic information facilitated by RNA-Seq is worth the increased computation over microarrays, specifically at three levels: absolute expression levels, differentially expressed genes identification, and expression QTL (eQTL) mapping in regions of the human brain. Using the United Kingdom Brain Expression Consortium (UKBEC) dataset, there is high agreement of gene expression levels measured by microarrays and RNA-seq when quantifying absolute expression levels and when identifying differentially expressed genes. These findings suggest that depending on the aims of a study, the relative ease of working with microarray data may outweigh the computational time and costs of RNA-Seq pipelines. On the other, there was low agreement when mapping eQTLs. However, a number of eQTLs associated with genes that play important roles in the brain were found in both platforms. For example, a trans-eQTL was mapped that is associated with the MPZ gene in the substantia nigra. These eQTLs that we have highlighted are extremely promising candidates that merit further investigation.
 分享
分享




 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
