In deep bioinformatic characterization of a novel fumarate hydratase variant FH c.199T > G; (p.Tyr67Asp) in hereditary leiomyomatosis and renal cell carcinoma.
Anisse Chami, Thalía Rodrigues de Souza Zózimo, Thamiris Matias Alves, Carolina Guimarães Ramos Matosinho, Cleydson Santos, Marcela Mattos Simões, Walter Luiz Ribeiro Cabral, Bernardo Ferreira de Paula Ricardo, Agnaldo Lopes da Silva Filho, Maria Raquel Santos Carvalho, Letícia da Conceição Braga
{"title":"In deep bioinformatic characterization of a novel fumarate hydratase variant FH c.199T > G; (p.Tyr67Asp) in hereditary leiomyomatosis and renal cell carcinoma.","authors":"Anisse Chami, Thalía Rodrigues de Souza Zózimo, Thamiris Matias Alves, Carolina Guimarães Ramos Matosinho, Cleydson Santos, Marcela Mattos Simões, Walter Luiz Ribeiro Cabral, Bernardo Ferreira de Paula Ricardo, Agnaldo Lopes da Silva Filho, Maria Raquel Santos Carvalho, Letícia da Conceição Braga","doi":"10.1007/s10689-023-00335-2","DOIUrl":null,"url":null,"abstract":"<p><p>Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is a rare, autosomal dominant tumor predisposition syndrome characterized by variable development of multiple skin and uterus leiomyomas and aggressive forms of renal cell carcinoma (RCC). Mutations in fumarate hydratase (FH), one of the proteins in homologous recombination repair, precede the development of HLRCC with high penetrance. Considering the risk of early metastasis of RCC, FH has been included in mutation screening panels. The identification of a pathogenic FH variant guides the screening for tumors in the carriers. However, variants of uncertain significance (VUS) are frequent findings, limiting the clinical value of the mutation screening. Here, we describe the associated phenotype and an in-depth, multi-step Bioinformatic evaluation of the germline FH c.199T > G (p.Tyr67 > Asp) variant segregated in an HLRCC family. Evidence for FH c.199T > G; (p.Tyr67Asp) pathogenicity includes the variant segregation with the disease in three affected family members, its absence in populational databases, and the deep evolutionary conservation of the Tyr67 residue. At the protein level, this residue substitution causes the loss of molecular bonds and ionic interactions, affecting molecular dynamics and protein stability. Considering ACMG/AMP criteria, we propose the reclassification of the FH c.199T > G; (p.Tyr67Asp) variant to \"likely pathogenic\". In addition, the in-depth, in silico approach used here allowed us to understand how and why FH c.199T > G; (p.Tyr67Asp) could cause HLRCC. This could help in clinical management decisions concerning the monitoring of unaffected family members having this variant.</p>","PeriodicalId":12336,"journal":{"name":"Familial Cancer","volume":" ","pages":"481-486"},"PeriodicalIF":2.0000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Familial Cancer","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10689-023-00335-2","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/15 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
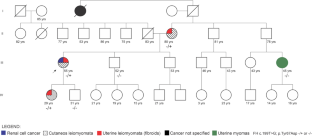
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is a rare, autosomal dominant tumor predisposition syndrome characterized by variable development of multiple skin and uterus leiomyomas and aggressive forms of renal cell carcinoma (RCC). Mutations in fumarate hydratase (FH), one of the proteins in homologous recombination repair, precede the development of HLRCC with high penetrance. Considering the risk of early metastasis of RCC, FH has been included in mutation screening panels. The identification of a pathogenic FH variant guides the screening for tumors in the carriers. However, variants of uncertain significance (VUS) are frequent findings, limiting the clinical value of the mutation screening. Here, we describe the associated phenotype and an in-depth, multi-step Bioinformatic evaluation of the germline FH c.199T > G (p.Tyr67 > Asp) variant segregated in an HLRCC family. Evidence for FH c.199T > G; (p.Tyr67Asp) pathogenicity includes the variant segregation with the disease in three affected family members, its absence in populational databases, and the deep evolutionary conservation of the Tyr67 residue. At the protein level, this residue substitution causes the loss of molecular bonds and ionic interactions, affecting molecular dynamics and protein stability. Considering ACMG/AMP criteria, we propose the reclassification of the FH c.199T > G; (p.Tyr67Asp) variant to "likely pathogenic". In addition, the in-depth, in silico approach used here allowed us to understand how and why FH c.199T > G; (p.Tyr67Asp) could cause HLRCC. This could help in clinical management decisions concerning the monitoring of unaffected family members having this variant.
期刊介绍:
In recent years clinical cancer genetics has become increasingly important. Several events, in particular the developments in DNA-based technology, have contributed to this evolution. Clinical cancer genetics has now matured to a medical discipline which is truly multidisciplinary in which clinical and molecular geneticists work together with clinical and medical oncologists as well as with psycho-social workers.
Due to the multidisciplinary nature of clinical cancer genetics most papers are currently being published in a wide variety of journals on epidemiology, oncology and genetics. Familial Cancer provides a forum bringing these topics together focusing on the interests and needs of the clinician.
The journal mainly concentrates on clinical cancer genetics. Most major areas in the field shall be included, such as epidemiology of familial cancer, molecular analysis and diagnosis, clinical expression, treatment and prevention, counselling and the health economics of familial cancer.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
