Carolina Isabela Mucellini, José Valter Joaquim Silva Júnior, Pablo Sebastian Britto de Oliveira, Rudi Weiblen, Eduardo Furtado Flores
{"title":"Novel genomic targets for proper subtyping of bovine viral diarrhea virus 1 (BVDV-1) and BVDV-2.","authors":"Carolina Isabela Mucellini, José Valter Joaquim Silva Júnior, Pablo Sebastian Britto de Oliveira, Rudi Weiblen, Eduardo Furtado Flores","doi":"10.1007/s11262-023-02022-x","DOIUrl":null,"url":null,"abstract":"<p><p>Whole-genome phylogenetic analysis, the most suitable strategy for subtyping bovine viral diarrhea virus 1 (BVDV-1) and BVDV-2, is not feasible for many laboratories. Consequently, BVDV isolates/strains have been frequently subtyped based on analysis of single genomic regions, mainly the 5' untranslated region (UTR). This approach, however, may lead to inaccurate and/or poorly statistically supported viral classification. Herein, we describe novel primer sets whose amplicons may be easily sequenced and used for BVDV subtyping. Initially, genomic regions previously described as the most suitable targets for BVDV subtyping were analyzed for design of high-coverage primers. The putative amplicons were analyzed in silico for their suitability to reproduce the phylogenetic classification of 118 BVDV-1 and 88 BVDV-2 complete/near-complete genomes (CNCGs) (GenBank). This analysis was also performed considering the region amplifiable by primers HCV90-368, 324-326 and BP189-389 (5'UTR), which have been used for BVDV diagnosis and/or classification. After confirming the agreement between the analyses of our primers' amplicon versus the CNCGs, we optimized the RT-PCRs and evaluated their performance for amplification of BVDV isolates/strains (n = 35 for BVDV-1; n = 33 for BVDV-2). Among the potential targets for BVDV subtyping, we designed high-coverage primers for NS3-NS4A (BVDV-1) (526 bp amplicon) and NS5B (BVDV-2) (728 bp). The classification based on these regions fully reproduced the subtyping of all CNCGs. On the other hand, subtyping based on the putative amplicons from primers HCV90-368, 324-326 and BP189-389 showed disagreements in relation the CNCG analysis. The NS3-NS4A and NS5B primers also allowed the amplification of all BVDV isolates/strains tested. Finally, we suggest the use of these primers in future phylogenetic and epidemiological studies of BVDVs.</p>","PeriodicalId":51212,"journal":{"name":"Virus Genes","volume":" ","pages":"836-844"},"PeriodicalIF":1.9000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Virus Genes","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11262-023-02022-x","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/17 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
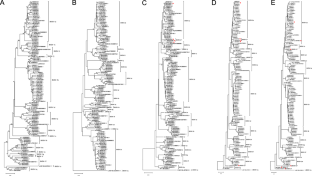
Whole-genome phylogenetic analysis, the most suitable strategy for subtyping bovine viral diarrhea virus 1 (BVDV-1) and BVDV-2, is not feasible for many laboratories. Consequently, BVDV isolates/strains have been frequently subtyped based on analysis of single genomic regions, mainly the 5' untranslated region (UTR). This approach, however, may lead to inaccurate and/or poorly statistically supported viral classification. Herein, we describe novel primer sets whose amplicons may be easily sequenced and used for BVDV subtyping. Initially, genomic regions previously described as the most suitable targets for BVDV subtyping were analyzed for design of high-coverage primers. The putative amplicons were analyzed in silico for their suitability to reproduce the phylogenetic classification of 118 BVDV-1 and 88 BVDV-2 complete/near-complete genomes (CNCGs) (GenBank). This analysis was also performed considering the region amplifiable by primers HCV90-368, 324-326 and BP189-389 (5'UTR), which have been used for BVDV diagnosis and/or classification. After confirming the agreement between the analyses of our primers' amplicon versus the CNCGs, we optimized the RT-PCRs and evaluated their performance for amplification of BVDV isolates/strains (n = 35 for BVDV-1; n = 33 for BVDV-2). Among the potential targets for BVDV subtyping, we designed high-coverage primers for NS3-NS4A (BVDV-1) (526 bp amplicon) and NS5B (BVDV-2) (728 bp). The classification based on these regions fully reproduced the subtyping of all CNCGs. On the other hand, subtyping based on the putative amplicons from primers HCV90-368, 324-326 and BP189-389 showed disagreements in relation the CNCG analysis. The NS3-NS4A and NS5B primers also allowed the amplification of all BVDV isolates/strains tested. Finally, we suggest the use of these primers in future phylogenetic and epidemiological studies of BVDVs.
期刊介绍:
Viruses are convenient models for the elucidation of life processes. The study of viruses is again on the cutting edge of biological sciences: systems biology, genomics, proteomics, metagenomics, using the newest most powerful tools.
Huge amounts of new details on virus interactions with the cell, other pathogens and the hosts – animal (including human), insect, fungal, plant, bacterial, and archaeal - and their role in infection and disease are forthcoming in perplexing details requiring analysis and comments.
Virus Genes is dedicated to the publication of studies on the structure and function of viruses and their genes, the molecular and systems interactions with the host and all applications derived thereof, providing a forum for the analysis of data and discussion of its implications, and the development of new hypotheses.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
