Takeaki Taniguchi, Miki Okuno, Takahiro Shinoda, Fumiya Kobayashi, Kazuki Takahashi, Hideaki Yuasa, Yuta Nakamura, Hiroyuki Tanaka, Rei Kajitani, Takehiko Itoh
{"title":"GINGER: an integrated method for high-accuracy prediction of gene structure in higher eukaryotes at the gene and exon level.","authors":"Takeaki Taniguchi, Miki Okuno, Takahiro Shinoda, Fumiya Kobayashi, Kazuki Takahashi, Hideaki Yuasa, Yuta Nakamura, Hiroyuki Tanaka, Rei Kajitani, Takehiko Itoh","doi":"10.1093/dnares/dsad017","DOIUrl":null,"url":null,"abstract":"<p><p>The prediction of gene structure within the genome sequence is the starting point of genome analysis, and its accuracy has a significant impact on the quality of subsequent analyses. Gene structure prediction is roughly divided into RNA-Seq-based methods, ab initio-based methods, homology-based methods, and the integration of individual prediction methods. Integrated methods are mainstream in recent genome projects because they improve prediction accuracy by combining or taking the best individual prediction findings; however, adequate prediction accuracy for eukaryotic species has not yet been achieved. Therefore, we developed an integrated tool, GINGER, that solves various issues related to gene structure prediction in higher eukaryotes. By handling artefacts in alignments of RNA and protein sequences, reconstructing gene structures via dynamic programming with appropriately weighted and scored exon/intron/intergenic regions, and applying different prediction processes and filtering criteria to multi-exon and single-exon genes, we achieved a significant improvement in accuracy compared to the existing integration methods. The feature of GINGER is its high prediction accuracy at the gene and exon levels, which is pronounced for species with more complex gene architectures. GINGER is implemented using Nextflow, which allows for the efficient and effective use of computing resources.</p>","PeriodicalId":51014,"journal":{"name":"DNA Research","volume":"30 4","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/cb/45/dsad017.PMC10439787.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"DNA Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/dnares/dsad017","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
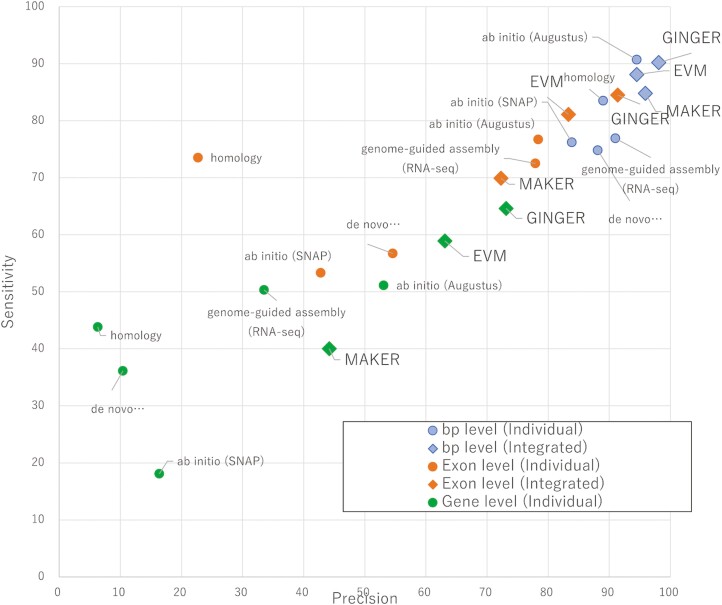
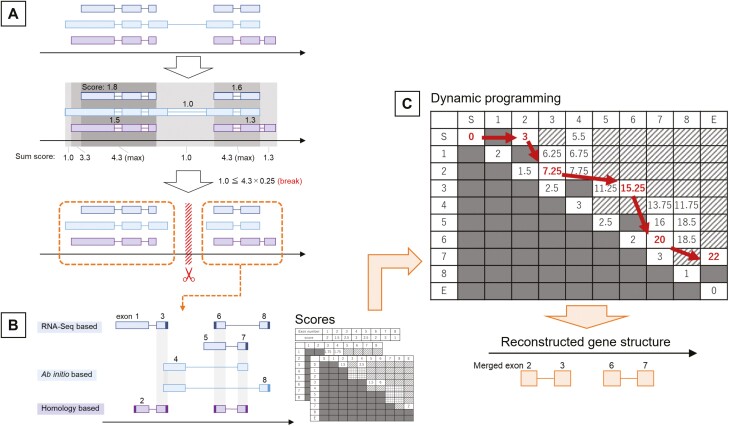
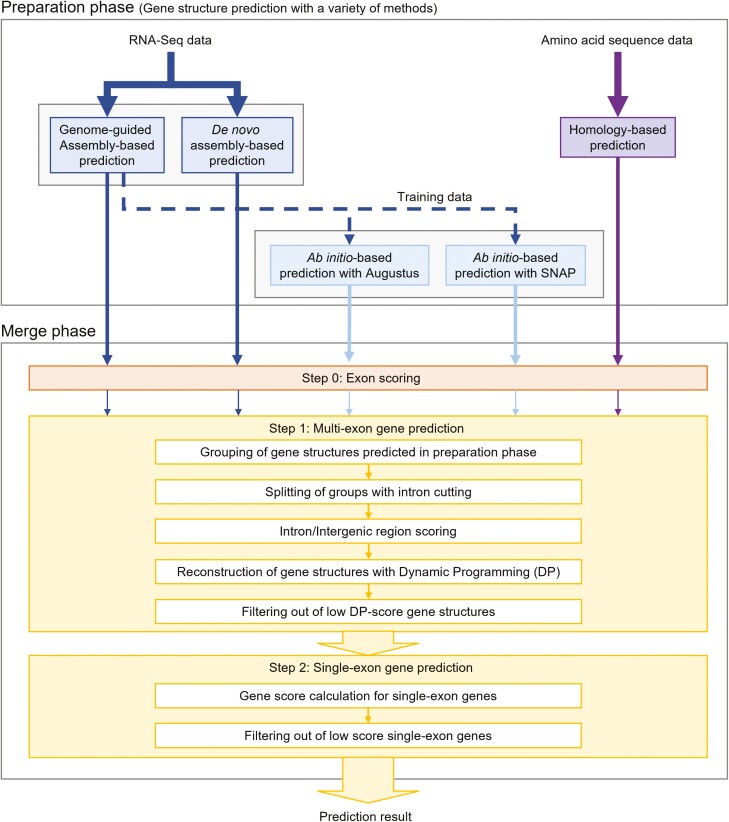
The prediction of gene structure within the genome sequence is the starting point of genome analysis, and its accuracy has a significant impact on the quality of subsequent analyses. Gene structure prediction is roughly divided into RNA-Seq-based methods, ab initio-based methods, homology-based methods, and the integration of individual prediction methods. Integrated methods are mainstream in recent genome projects because they improve prediction accuracy by combining or taking the best individual prediction findings; however, adequate prediction accuracy for eukaryotic species has not yet been achieved. Therefore, we developed an integrated tool, GINGER, that solves various issues related to gene structure prediction in higher eukaryotes. By handling artefacts in alignments of RNA and protein sequences, reconstructing gene structures via dynamic programming with appropriately weighted and scored exon/intron/intergenic regions, and applying different prediction processes and filtering criteria to multi-exon and single-exon genes, we achieved a significant improvement in accuracy compared to the existing integration methods. The feature of GINGER is its high prediction accuracy at the gene and exon levels, which is pronounced for species with more complex gene architectures. GINGER is implemented using Nextflow, which allows for the efficient and effective use of computing resources.
期刊介绍:
DNA Research is an internationally peer-reviewed journal which aims at publishing papers of highest quality in broad aspects of DNA and genome-related research. Emphasis will be made on the following subjects: 1) Sequencing and characterization of genomes/important genomic regions, 2) Comprehensive analysis of the functions of genes, gene families and genomes, 3) Techniques and equipments useful for structural and functional analysis of genes, gene families and genomes, 4) Computer algorithms and/or their applications relevant to structural and functional analysis of genes and genomes. The journal also welcomes novel findings in other scientific disciplines related to genomes.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
