Alexandra Unapanta, Farbod Shavarebi, Jacob Porath, Yiyi Shen, Carson Balen, Albert Nguyen, Josh Tseng, Weng Si Leong, Michelle Liu, Pawel Lis, Santiago M Di Pietro, Annie Hiniker
{"title":"Endogenous Rab38 regulates LRRK2's membrane recruitment and substrate Rab phosphorylation in melanocytes.","authors":"Alexandra Unapanta, Farbod Shavarebi, Jacob Porath, Yiyi Shen, Carson Balen, Albert Nguyen, Josh Tseng, Weng Si Leong, Michelle Liu, Pawel Lis, Santiago M Di Pietro, Annie Hiniker","doi":"10.1016/j.jbc.2023.105192","DOIUrl":null,"url":null,"abstract":"<p><p>Point mutations in leucine-rich repeat kinase 2 (LRRK2) cause Parkinson's disease and augment LRRK2's kinase activity. However, cellular pathways that endogenously enhance LRRK2 kinase function have not been identified. While overexpressed Rab29 draws LRRK2 to Golgi membranes to increase LRRK2 kinase activity, there is little evidence that endogenous Rab29 performs this function under physiological conditions. Here, we identify Rab38 as a novel physiologic regulator of LRRK2 in melanocytes. In mouse melanocytes, which express high levels of Rab38, Rab32, and Rab29, knockdown (or CRISPR knockout) of Rab38, but not Rab32 or Rab29, decreases phosphorylation of multiple LRRK2 substrates, including Rab10 and Rab12, by both endogenous LRRK2 and exogenous Parkinson's disease-mutant LRRK2. In B16-F10 mouse melanoma cells, Rab38 drives LRRK2 membrane association and overexpressed kinase-active LRRK2 shows striking pericentriolar recruitment, which is dependent on the presence of endogenous Rab38 but not Rab32 or Rab29. Consistently, knockdown or mutation of BLOC-3, the guanine nucleotide exchange factor for Rab38 and Rab32, inhibits Rab38's regulation of LRRK2. Deletion or mutation of LRRK2's Rab38-binding site in the N-terminal armadillo domain decreases LRRK2 membrane association, pericentriolar recruitment, and ability to phosphorylate Rab10. In sum, our data identify Rab38 as a physiologic regulator of LRRK2 function and lend support to a model in which LRRK2 plays a central role in Rab GTPase coordination of vesicular trafficking.</p>","PeriodicalId":22621,"journal":{"name":"The Journal of Biological Chemistry","volume":" ","pages":"105192"},"PeriodicalIF":0.0000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10551901/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Biological Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.jbc.2023.105192","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/23 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
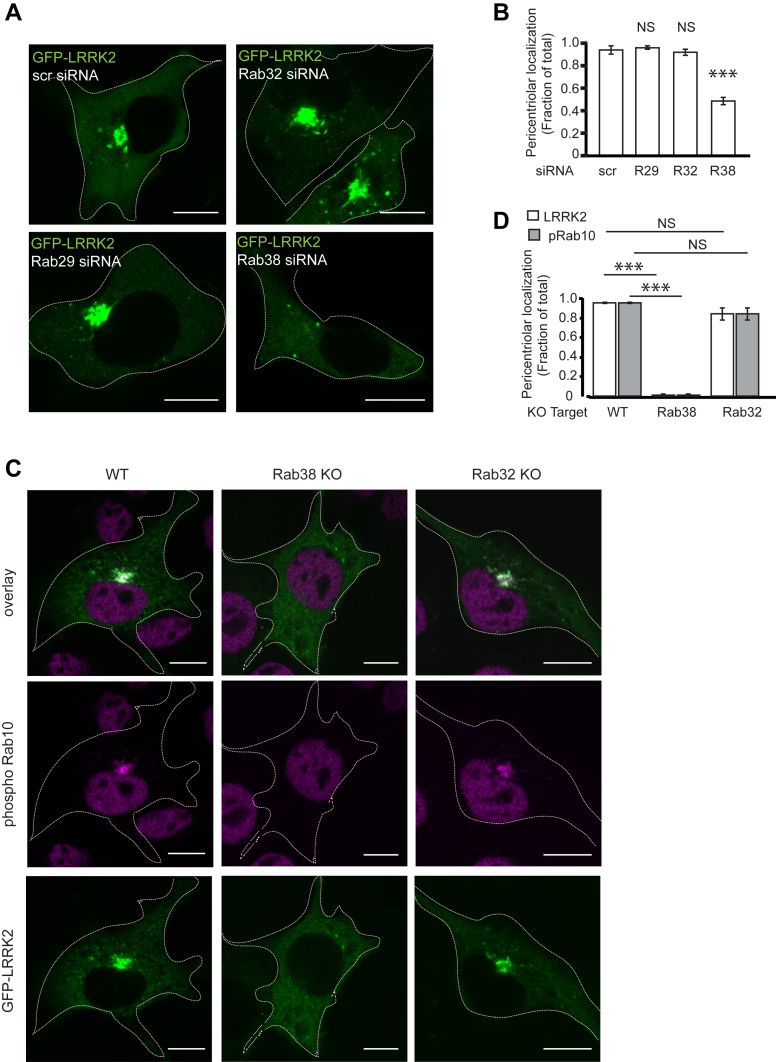
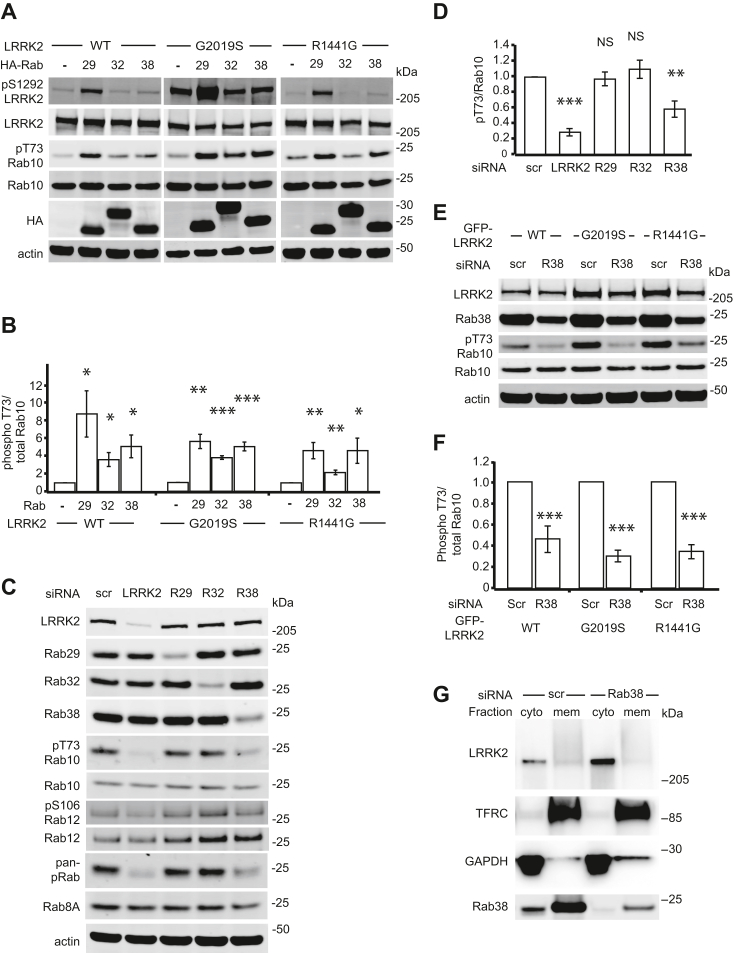
Point mutations in leucine-rich repeat kinase 2 (LRRK2) cause Parkinson's disease and augment LRRK2's kinase activity. However, cellular pathways that endogenously enhance LRRK2 kinase function have not been identified. While overexpressed Rab29 draws LRRK2 to Golgi membranes to increase LRRK2 kinase activity, there is little evidence that endogenous Rab29 performs this function under physiological conditions. Here, we identify Rab38 as a novel physiologic regulator of LRRK2 in melanocytes. In mouse melanocytes, which express high levels of Rab38, Rab32, and Rab29, knockdown (or CRISPR knockout) of Rab38, but not Rab32 or Rab29, decreases phosphorylation of multiple LRRK2 substrates, including Rab10 and Rab12, by both endogenous LRRK2 and exogenous Parkinson's disease-mutant LRRK2. In B16-F10 mouse melanoma cells, Rab38 drives LRRK2 membrane association and overexpressed kinase-active LRRK2 shows striking pericentriolar recruitment, which is dependent on the presence of endogenous Rab38 but not Rab32 or Rab29. Consistently, knockdown or mutation of BLOC-3, the guanine nucleotide exchange factor for Rab38 and Rab32, inhibits Rab38's regulation of LRRK2. Deletion or mutation of LRRK2's Rab38-binding site in the N-terminal armadillo domain decreases LRRK2 membrane association, pericentriolar recruitment, and ability to phosphorylate Rab10. In sum, our data identify Rab38 as a physiologic regulator of LRRK2 function and lend support to a model in which LRRK2 plays a central role in Rab GTPase coordination of vesicular trafficking.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
