{"title":"Block Aligner: an adaptive SIMD-accelerated aligner for sequences and position-specific scoring matrices.","authors":"Daniel Liu, Martin Steinegger","doi":"10.1093/bioinformatics/btad487","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Efficiently aligning sequences is a fundamental problem in bioinformatics. Many recent algorithms for computing alignments through Smith-Waterman-Gotoh dynamic programming (DP) exploit Single Instruction Multiple Data (SIMD) operations on modern CPUs for speed. However, these advances have largely ignored difficulties associated with efficiently handling complex scoring matrices or large gaps (insertions or deletions).</p><p><strong>Results: </strong>We propose a new SIMD-accelerated algorithm called Block Aligner for aligning nucleotide and protein sequences against other sequences or position-specific scoring matrices. We introduce a new paradigm that uses blocks in the DP matrix that greedily shift, grow, and shrink. This approach allows regions of the DP matrix to be adaptively computed. Our algorithm reaches over 5-10 times faster than some previous methods while incurring an error rate of less than 3% on protein and long read datasets, despite large gaps and low sequence identities.</p><p><strong>Availability and implementation: </strong>Our algorithm is implemented for global, local, and X-drop alignments. It is available as a Rust library (with C bindings) at https://github.com/Daniel-Liu-c0deb0t/block-aligner.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 8","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10457662/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad487","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Efficiently aligning sequences is a fundamental problem in bioinformatics. Many recent algorithms for computing alignments through Smith-Waterman-Gotoh dynamic programming (DP) exploit Single Instruction Multiple Data (SIMD) operations on modern CPUs for speed. However, these advances have largely ignored difficulties associated with efficiently handling complex scoring matrices or large gaps (insertions or deletions).
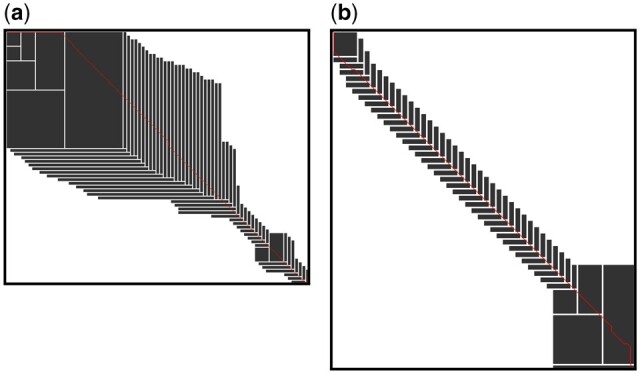
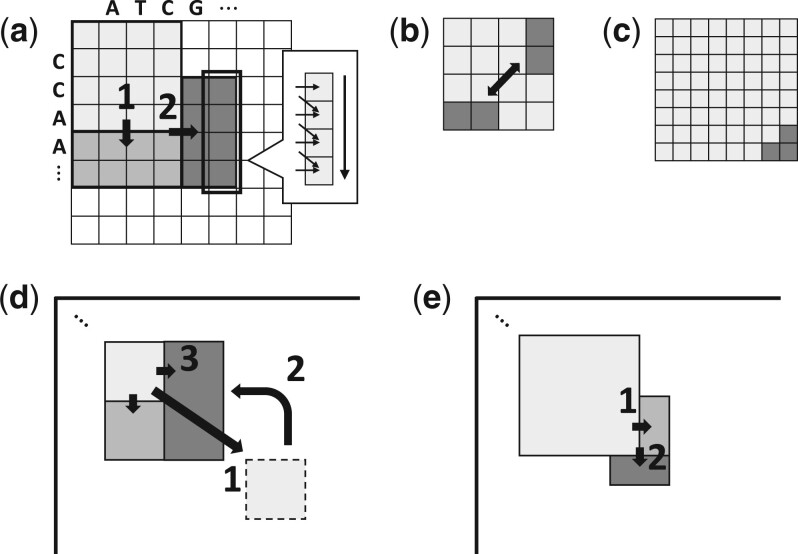
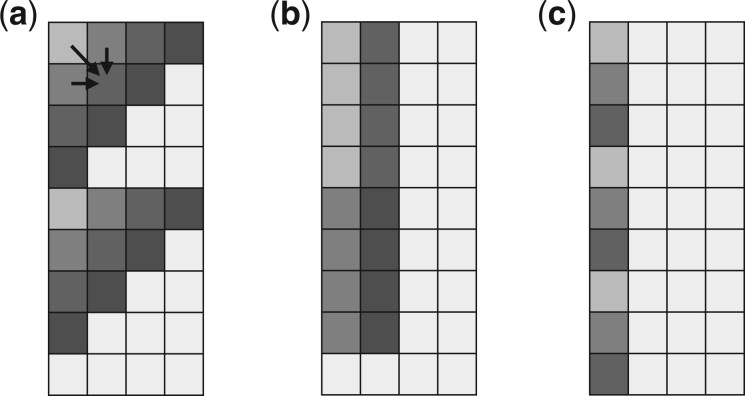
Results: We propose a new SIMD-accelerated algorithm called Block Aligner for aligning nucleotide and protein sequences against other sequences or position-specific scoring matrices. We introduce a new paradigm that uses blocks in the DP matrix that greedily shift, grow, and shrink. This approach allows regions of the DP matrix to be adaptively computed. Our algorithm reaches over 5-10 times faster than some previous methods while incurring an error rate of less than 3% on protein and long read datasets, despite large gaps and low sequence identities.
Availability and implementation: Our algorithm is implemented for global, local, and X-drop alignments. It is available as a Rust library (with C bindings) at https://github.com/Daniel-Liu-c0deb0t/block-aligner.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
