{"title":"Flux balance analysis-based metabolic modeling of microbial secondary metabolism: Current status and outlook.","authors":"Sizhe Qiu, Aidong Yang, Hong Zeng","doi":"10.1371/journal.pcbi.1011391","DOIUrl":null,"url":null,"abstract":"<p><p>In microorganisms, different from primary metabolism for cellular growth, secondary metabolism is for ecological interactions and stress responses and an important source of natural products widely used in various areas such as pharmaceutics and food additives. With advancements of sequencing technologies and bioinformatics tools, a large number of biosynthetic gene clusters of secondary metabolites have been discovered from microbial genomes. However, due to challenges from the difficulty of genome-scale pathway reconstruction and the limitation of conventional flux balance analysis (FBA) on secondary metabolism, the quantitative modeling of secondary metabolism is poorly established, in contrast to that of primary metabolism. This review first discusses current efforts on the reconstruction of secondary metabolic pathways in genome-scale metabolic models (GSMMs), as well as related FBA-based modeling techniques. Additionally, potential extensions of FBA are suggested to improve the prediction accuracy of secondary metabolite production. As this review posits, biosynthetic pathway reconstruction for various secondary metabolites will become automated and a modeling framework capturing secondary metabolism onset will enhance the predictive power. Expectedly, an improved FBA-based modeling workflow will facilitate quantitative study of secondary metabolism and in silico design of engineering strategies for natural product production.</p>","PeriodicalId":49688,"journal":{"name":"PLoS Computational Biology","volume":"19 8","pages":"e1011391"},"PeriodicalIF":3.6000,"publicationDate":"2023-08-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10449171/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Computational Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pcbi.1011391","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
In microorganisms, different from primary metabolism for cellular growth, secondary metabolism is for ecological interactions and stress responses and an important source of natural products widely used in various areas such as pharmaceutics and food additives. With advancements of sequencing technologies and bioinformatics tools, a large number of biosynthetic gene clusters of secondary metabolites have been discovered from microbial genomes. However, due to challenges from the difficulty of genome-scale pathway reconstruction and the limitation of conventional flux balance analysis (FBA) on secondary metabolism, the quantitative modeling of secondary metabolism is poorly established, in contrast to that of primary metabolism. This review first discusses current efforts on the reconstruction of secondary metabolic pathways in genome-scale metabolic models (GSMMs), as well as related FBA-based modeling techniques. Additionally, potential extensions of FBA are suggested to improve the prediction accuracy of secondary metabolite production. As this review posits, biosynthetic pathway reconstruction for various secondary metabolites will become automated and a modeling framework capturing secondary metabolism onset will enhance the predictive power. Expectedly, an improved FBA-based modeling workflow will facilitate quantitative study of secondary metabolism and in silico design of engineering strategies for natural product production.
期刊介绍:
PLOS Computational Biology features works of exceptional significance that further our understanding of living systems at all scales—from molecules and cells, to patient populations and ecosystems—through the application of computational methods. Readers include life and computational scientists, who can take the important findings presented here to the next level of discovery.
Research articles must be declared as belonging to a relevant section. More information about the sections can be found in the submission guidelines.
Research articles should model aspects of biological systems, demonstrate both methodological and scientific novelty, and provide profound new biological insights.
Generally, reliability and significance of biological discovery through computation should be validated and enriched by experimental studies. Inclusion of experimental validation is not required for publication, but should be referenced where possible. Inclusion of experimental validation of a modest biological discovery through computation does not render a manuscript suitable for PLOS Computational Biology.
Research articles specifically designated as Methods papers should describe outstanding methods of exceptional importance that have been shown, or have the promise to provide new biological insights. The method must already be widely adopted, or have the promise of wide adoption by a broad community of users. Enhancements to existing published methods will only be considered if those enhancements bring exceptional new capabilities.

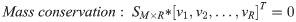
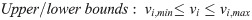



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
