Introduction of a multiplex amplicon sequencing assay to quantify DNA methylation in target cytosine markers underlying four selected epigenetic clocks.
Ewelina Pośpiech, Aleksandra Pisarek, Joanna Rudnicka, Rezvan Noroozi, Michał Boroń, Aleksander Masny, Bożena Wysocka, Kamila Migacz-Gruszka, Dagmara Lisman, Paulina Pruszkowska-Przybylska, Magdalena Kobus, Maria Szargut, Joanna Dowejko, Kamila Stanisz, Julia Zacharczuk, Piotr Zieliński, Aneta Sitek, Andrzej Ossowski, Magdalena Spólnicka, Wojciech Branicki
{"title":"Introduction of a multiplex amplicon sequencing assay to quantify DNA methylation in target cytosine markers underlying four selected epigenetic clocks.","authors":"Ewelina Pośpiech, Aleksandra Pisarek, Joanna Rudnicka, Rezvan Noroozi, Michał Boroń, Aleksander Masny, Bożena Wysocka, Kamila Migacz-Gruszka, Dagmara Lisman, Paulina Pruszkowska-Przybylska, Magdalena Kobus, Maria Szargut, Joanna Dowejko, Kamila Stanisz, Julia Zacharczuk, Piotr Zieliński, Aneta Sitek, Andrzej Ossowski, Magdalena Spólnicka, Wojciech Branicki","doi":"10.1186/s13148-023-01545-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>DNA methylation analysis has proven to be a powerful tool for age assessment. However, the implementation of epigenetic age prediction in diagnostics or routine forensic casework requires appropriate laboratory methods. In this study, we aimed to compare the performance of large-scale DNA methylation analysis protocols that show promise in terms of accuracy, throughput, multiplexing capacity, and high sensitivity.</p><p><strong>Results: </strong>The protocols were designed to target a predefined panel of 161 genomic CG/CA sites from four known estimators of epigenetic age-related parameters, optimized and validated using artificially methylated controls or blood samples. We successfully targeted 96% of these loci using two enrichment protocols: Ion AmpliSeq™, an amplicon-based method integrated with Ion Torrent S5, and SureSelect<sup>XT</sup> Methyl-Seq, a hybridization-based method followed by MiSeq FGx sequencing. Both protocols demonstrated high accuracy and robustness. Although hybridization assays have greater multiplexing capabilities, the best overall performance was observed for the amplicon-based protocol with the lowest variability in DNA methylation at 25 ng of starting DNA, mean observed marker coverage of ~ 6.7 k reads, and accuracy of methylation quantification with a mean absolute difference between observed and expected methylation beta value of 0.054. The Ion AmpliSeq method correlated strongly with genome-scale EPIC microarray data (R = 0.91) and showed superiority in terms of methylation measurement accuracy. Method-to-method bias was accounted for by the use of linear transformation, which provided a highly accurate prediction of calendar age with a mean absolute error of less than 5 years for the VISAGE and Hannum age clocks used. The pace of aging (PoAm) and the mortality risk score (MRS) estimators included in our panel represent next-generation clocks, were found to have low to moderate correlations with the VISAGE and Hannum models (R < 0.75), and thus may capture different aspects of epigenetic aging.</p><p><strong>Conclusions: </strong>We propose a laboratory tool that allows the quantification of DNA methylation in cytosines underlying four different clocks, thus providing broad information on epigenetic aging while maintaining a reasonable number of CpG markers, opening the way to a wide range of applications in forensics, medicine, and healthcare.</p>","PeriodicalId":48652,"journal":{"name":"Clinical Epigenetics","volume":"15 1","pages":"128"},"PeriodicalIF":4.4000,"publicationDate":"2023-08-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10416531/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-023-01545-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
Background: DNA methylation analysis has proven to be a powerful tool for age assessment. However, the implementation of epigenetic age prediction in diagnostics or routine forensic casework requires appropriate laboratory methods. In this study, we aimed to compare the performance of large-scale DNA methylation analysis protocols that show promise in terms of accuracy, throughput, multiplexing capacity, and high sensitivity.
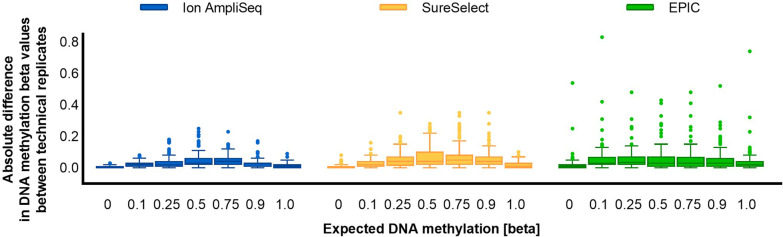
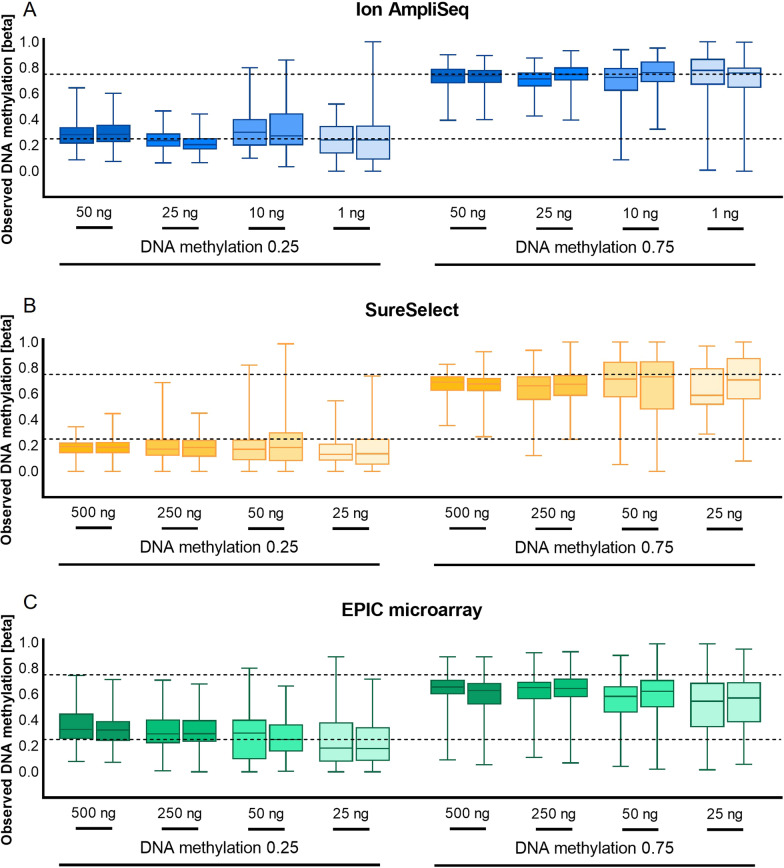
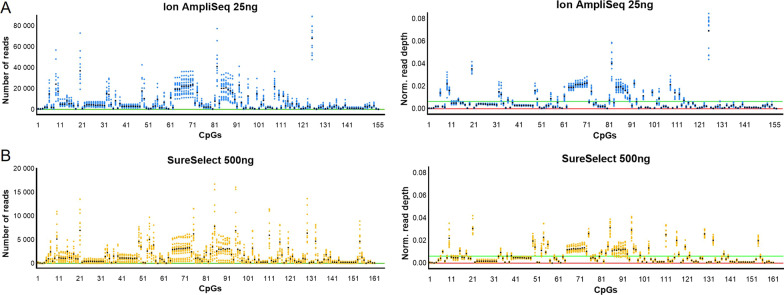
Results: The protocols were designed to target a predefined panel of 161 genomic CG/CA sites from four known estimators of epigenetic age-related parameters, optimized and validated using artificially methylated controls or blood samples. We successfully targeted 96% of these loci using two enrichment protocols: Ion AmpliSeq™, an amplicon-based method integrated with Ion Torrent S5, and SureSelectXT Methyl-Seq, a hybridization-based method followed by MiSeq FGx sequencing. Both protocols demonstrated high accuracy and robustness. Although hybridization assays have greater multiplexing capabilities, the best overall performance was observed for the amplicon-based protocol with the lowest variability in DNA methylation at 25 ng of starting DNA, mean observed marker coverage of ~ 6.7 k reads, and accuracy of methylation quantification with a mean absolute difference between observed and expected methylation beta value of 0.054. The Ion AmpliSeq method correlated strongly with genome-scale EPIC microarray data (R = 0.91) and showed superiority in terms of methylation measurement accuracy. Method-to-method bias was accounted for by the use of linear transformation, which provided a highly accurate prediction of calendar age with a mean absolute error of less than 5 years for the VISAGE and Hannum age clocks used. The pace of aging (PoAm) and the mortality risk score (MRS) estimators included in our panel represent next-generation clocks, were found to have low to moderate correlations with the VISAGE and Hannum models (R < 0.75), and thus may capture different aspects of epigenetic aging.
Conclusions: We propose a laboratory tool that allows the quantification of DNA methylation in cytosines underlying four different clocks, thus providing broad information on epigenetic aging while maintaining a reasonable number of CpG markers, opening the way to a wide range of applications in forensics, medicine, and healthcare.
Clinical EpigeneticsBiochemistry, Genetics and Molecular Biology-Developmental Biology
CiteScore
8.90
自引率
5.30%
发文量
150
审稿时长
12 weeks
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
