Ping Wang, Jinrong Liu, Xiaojuan Tan, Fred Yang, James McCabe, Jay Zhang
{"title":"Pharmacokinetics and Drug-Drug Interaction of Ocedurenone (KBP-5074) in vitro and in vivo.","authors":"Ping Wang, Jinrong Liu, Xiaojuan Tan, Fred Yang, James McCabe, Jay Zhang","doi":"10.1007/s13318-023-00837-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Ocedurenone (KBP-5074) is a novel nonsteroidal mineralocorticoid receptor antagonist that has demonstrated safety and efficacy in clinical trials in patients with uncontrolled hypertension and stage 3b/4 chronic kidney disease. This study evaluated the involvement of cytochrome P450 (CYP) isozymes and drug transporters in the biotransformation of ocedurenone, and whether ocedurenone inhibited or induced CYP enzymes and transporters. Clinical pharmacokinetic drug-drug interaction (DDI) of ocedurenone with CYP3A inhibitor and inducer were investigated in healthy volunteers.</p><p><strong>Methods: </strong>In vitro tests were conducted to determine which CYP enzymes were involved in ocedurenone's metabolism and whether ocedurenone inhibited or induced these CYP enzymes; ocedurenone substrate characteristics for efflux and uptake transporters and its inhibitory potential on major drug transporters were also assessed. A clinical DDI study was conducted in healthy volunteers to evaluate the effects of a strong CYP3A inhibitor (itraconazole) and inducer (rifampin) on ocedurenone's pharmacokinetics.</p><p><strong>Results: </strong>The in vitro study showed that ocedurenone was primarily metabolized by CYP3A4 and that it did not inhibit CYP enzymes. Ocedurenone appeared to be a substrate of BCRP and P-gp efflux transporters and inhibited BCRP, BSEP, MDR1, MATE1 and 2-K, OATP1B1/3, and OCT1. The clinical DDI study showed that itraconazole reduced ocedurenone's oral clearance by 51% and increased area under the plasma concentration-time curve extrapolated to infinity (AUC<sub>0-inf</sub>) by 104%, while rifampin increased its oral clearance by 6.4-fold and decreased plasma AUC<sub>0-inf</sub> by 84%.</p><p><strong>Conclusion: </strong>Ocedurenone was shown to be a CYP3A substrate, with no inhibition potential on major drug metabolizing CYP enzymes and transporters at clinical efficacious doses. Ocedurenone did not induce CYP1A2 and 3A4 activity in cultured human primary hepatocytes. Clinical DDI study indicated ocedurenone was well tolerated when administered as a single 0.5-mg dose both alone and with itraconazole or rifampin, and while itraconazole had a weak effect on ocedurenone's pharmacokinetics, rifampin had a significant effect reducing systemic exposures.</p>","PeriodicalId":11939,"journal":{"name":"European Journal of Drug Metabolism and Pharmacokinetics","volume":"48 4","pages":"397-410"},"PeriodicalIF":2.4000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7a/3f/13318_2023_Article_837.PMC10322960.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Drug Metabolism and Pharmacokinetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s13318-023-00837-5","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 1
Abstract
Background and objectives: Ocedurenone (KBP-5074) is a novel nonsteroidal mineralocorticoid receptor antagonist that has demonstrated safety and efficacy in clinical trials in patients with uncontrolled hypertension and stage 3b/4 chronic kidney disease. This study evaluated the involvement of cytochrome P450 (CYP) isozymes and drug transporters in the biotransformation of ocedurenone, and whether ocedurenone inhibited or induced CYP enzymes and transporters. Clinical pharmacokinetic drug-drug interaction (DDI) of ocedurenone with CYP3A inhibitor and inducer were investigated in healthy volunteers.
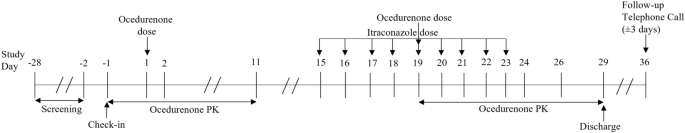
Methods: In vitro tests were conducted to determine which CYP enzymes were involved in ocedurenone's metabolism and whether ocedurenone inhibited or induced these CYP enzymes; ocedurenone substrate characteristics for efflux and uptake transporters and its inhibitory potential on major drug transporters were also assessed. A clinical DDI study was conducted in healthy volunteers to evaluate the effects of a strong CYP3A inhibitor (itraconazole) and inducer (rifampin) on ocedurenone's pharmacokinetics.
Results: The in vitro study showed that ocedurenone was primarily metabolized by CYP3A4 and that it did not inhibit CYP enzymes. Ocedurenone appeared to be a substrate of BCRP and P-gp efflux transporters and inhibited BCRP, BSEP, MDR1, MATE1 and 2-K, OATP1B1/3, and OCT1. The clinical DDI study showed that itraconazole reduced ocedurenone's oral clearance by 51% and increased area under the plasma concentration-time curve extrapolated to infinity (AUC0-inf) by 104%, while rifampin increased its oral clearance by 6.4-fold and decreased plasma AUC0-inf by 84%.
Conclusion: Ocedurenone was shown to be a CYP3A substrate, with no inhibition potential on major drug metabolizing CYP enzymes and transporters at clinical efficacious doses. Ocedurenone did not induce CYP1A2 and 3A4 activity in cultured human primary hepatocytes. Clinical DDI study indicated ocedurenone was well tolerated when administered as a single 0.5-mg dose both alone and with itraconazole or rifampin, and while itraconazole had a weak effect on ocedurenone's pharmacokinetics, rifampin had a significant effect reducing systemic exposures.
期刊介绍:
Hepatology International is a peer-reviewed journal featuring articles written by clinicians, clinical researchers and basic scientists is dedicated to research and patient care issues in hepatology. This journal focuses mainly on new and emerging diagnostic and treatment options, protocols and molecular and cellular basis of disease pathogenesis, new technologies, in liver and biliary sciences.
Hepatology International publishes original research articles related to clinical care and basic research; review articles; consensus guidelines for diagnosis and treatment; invited editorials, and controversies in contemporary issues. The journal does not publish case reports.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
