Yan Zhu, Lingling Zhao, Naifeng Wen, Junjie Wang, Chunyu Wang
{"title":"DataDTA: a multi-feature and dual-interaction aggregation framework for drug-target binding affinity prediction.","authors":"Yan Zhu, Lingling Zhao, Naifeng Wen, Junjie Wang, Chunyu Wang","doi":"10.1093/bioinformatics/btad560","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Accurate prediction of drug-target binding affinity (DTA) is crucial for drug discovery. The increase in the publication of large-scale DTA datasets enables the development of various computational methods for DTA prediction. Numerous deep learning-based methods have been proposed to predict affinities, some of which only utilize original sequence information or complex structures, but the effective combination of various information and protein-binding pockets have not been fully mined. Therefore, a new method that integrates available key information is urgently needed to predict DTA and accelerate the drug discovery process.</p><p><strong>Results: </strong>In this study, we propose a novel deep learning-based predictor termed DataDTA to estimate the affinities of drug-target pairs. DataDTA utilizes descriptors of predicted pockets and sequences of proteins, as well as low-dimensional molecular features and SMILES strings of compounds as inputs. Specifically, the pockets were predicted from the three-dimensional structure of proteins and their descriptors were extracted as the partial input features for DTA prediction. The molecular representation of compounds based on algebraic graph features was collected to supplement the input information of targets. Furthermore, to ensure effective learning of multiscale interaction features, a dual-interaction aggregation neural network strategy was developed. DataDTA was compared with state-of-the-art methods on different datasets, and the results showed that DataDTA is a reliable prediction tool for affinities estimation. Specifically, the concordance index (CI) of DataDTA is 0.806 and the Pearson correlation coefficient (R) value is 0.814 on the test dataset, which is higher than other methods.</p><p><strong>Availability and implementation: </strong>The codes and datasets of DataDTA are available at https://github.com/YanZhu06/DataDTA.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10516524/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad560","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Accurate prediction of drug-target binding affinity (DTA) is crucial for drug discovery. The increase in the publication of large-scale DTA datasets enables the development of various computational methods for DTA prediction. Numerous deep learning-based methods have been proposed to predict affinities, some of which only utilize original sequence information or complex structures, but the effective combination of various information and protein-binding pockets have not been fully mined. Therefore, a new method that integrates available key information is urgently needed to predict DTA and accelerate the drug discovery process.
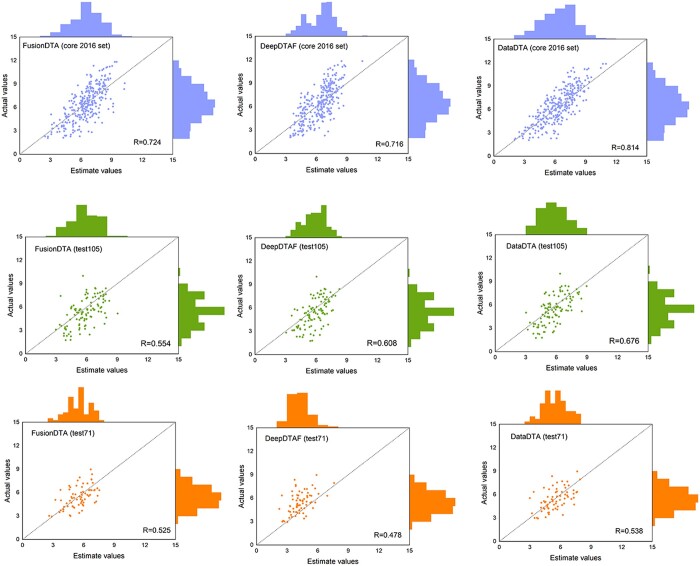
Results: In this study, we propose a novel deep learning-based predictor termed DataDTA to estimate the affinities of drug-target pairs. DataDTA utilizes descriptors of predicted pockets and sequences of proteins, as well as low-dimensional molecular features and SMILES strings of compounds as inputs. Specifically, the pockets were predicted from the three-dimensional structure of proteins and their descriptors were extracted as the partial input features for DTA prediction. The molecular representation of compounds based on algebraic graph features was collected to supplement the input information of targets. Furthermore, to ensure effective learning of multiscale interaction features, a dual-interaction aggregation neural network strategy was developed. DataDTA was compared with state-of-the-art methods on different datasets, and the results showed that DataDTA is a reliable prediction tool for affinities estimation. Specifically, the concordance index (CI) of DataDTA is 0.806 and the Pearson correlation coefficient (R) value is 0.814 on the test dataset, which is higher than other methods.
Availability and implementation: The codes and datasets of DataDTA are available at https://github.com/YanZhu06/DataDTA.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
