{"title":"A general framework for powerful confounder adjustment in omics association studies.","authors":"Asmita Roy, Jun Chen, Xianyang Zhang","doi":"10.1093/bioinformatics/btad563","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Genomic data are subject to various sources of confounding, such as demographic variables, biological heterogeneity, and batch effects. To identify genomic features associated with a variable of interest in the presence of confounders, the traditional approach involves fitting a confounder-adjusted regression model to each genomic feature, followed by multiplicity correction.</p><p><strong>Results: </strong>This study shows that the traditional approach is suboptimal and proposes a new two-dimensional false discovery rate control framework (2DFDR+) that provides significant power improvement over the conventional method and applies to a wide range of settings. 2DFDR+ uses marginal independence test statistics as auxiliary information to filter out less promising features, and FDR control is performed based on conditional independence test statistics in the remaining features. 2DFDR+ provides (asymptotically) valid inference from samples in settings where the conditional distribution of the genomic variables given the covariate of interest and the confounders is arbitrary and completely unknown. Promising finite sample performance is demonstrated via extensive simulations and real data applications.</p><p><strong>Availability and implementation: </strong>R codes and vignettes are available at https://github.com/asmita112358/tdfdr.np.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10539716/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad563","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Genomic data are subject to various sources of confounding, such as demographic variables, biological heterogeneity, and batch effects. To identify genomic features associated with a variable of interest in the presence of confounders, the traditional approach involves fitting a confounder-adjusted regression model to each genomic feature, followed by multiplicity correction.
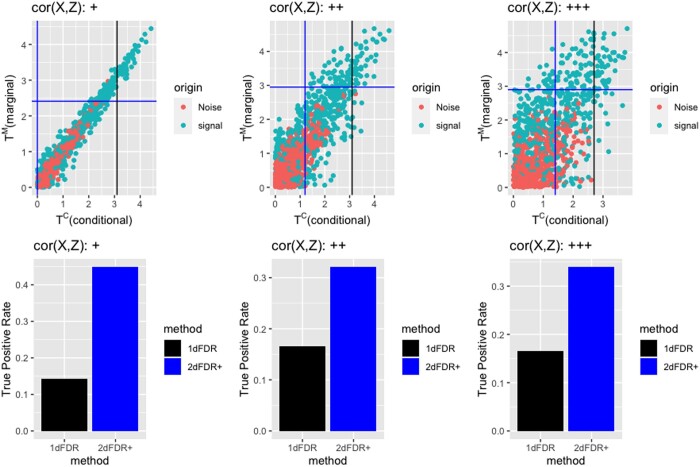
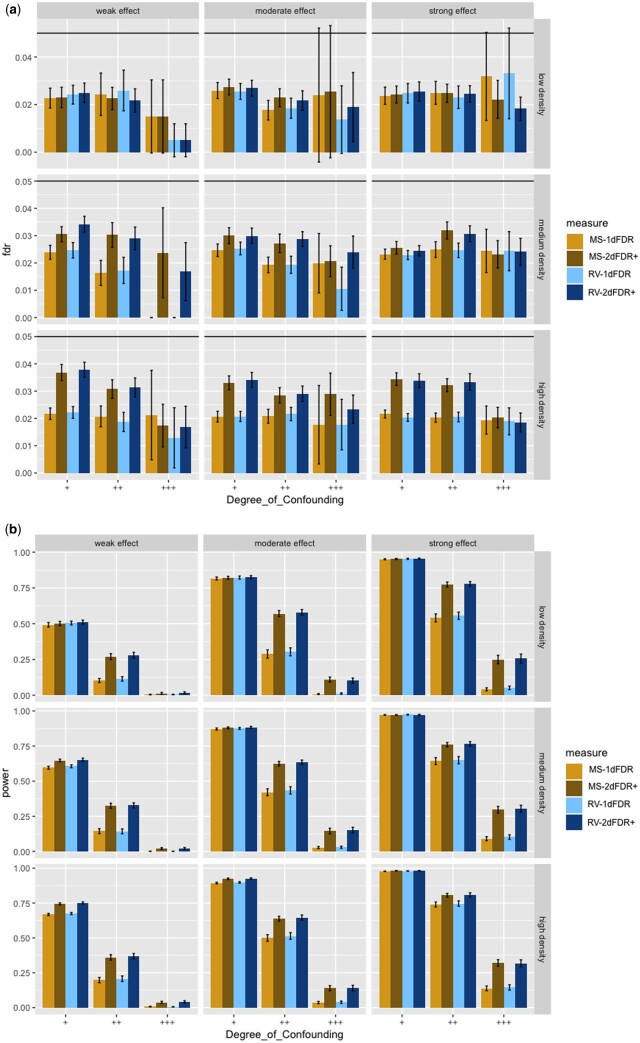
Results: This study shows that the traditional approach is suboptimal and proposes a new two-dimensional false discovery rate control framework (2DFDR+) that provides significant power improvement over the conventional method and applies to a wide range of settings. 2DFDR+ uses marginal independence test statistics as auxiliary information to filter out less promising features, and FDR control is performed based on conditional independence test statistics in the remaining features. 2DFDR+ provides (asymptotically) valid inference from samples in settings where the conditional distribution of the genomic variables given the covariate of interest and the confounders is arbitrary and completely unknown. Promising finite sample performance is demonstrated via extensive simulations and real data applications.
Availability and implementation: R codes and vignettes are available at https://github.com/asmita112358/tdfdr.np.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
