Katleen Janssens, Isabelle Neefs, Joe Ibrahim, Anne Schepers, Patrick Pauwels, Marc Peeters, Guy Van Camp, Ken Op de Beeck
{"title":"Epigenome-wide methylation analysis of colorectal carcinoma, adenoma and normal tissue reveals novel biomarkers addressing unmet clinical needs.","authors":"Katleen Janssens, Isabelle Neefs, Joe Ibrahim, Anne Schepers, Patrick Pauwels, Marc Peeters, Guy Van Camp, Ken Op de Beeck","doi":"10.1186/s13148-023-01516-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Biomarker discovery in colorectal cancer has mostly focused on methylation patterns in normal and colorectal tumor tissue, but adenomas remain understudied. Therefore, we performed the first epigenome-wide study to profile methylation of all three tissue types combined and to identify discriminatory biomarkers.</p><p><strong>Results: </strong>Public methylation array data (Illumina EPIC and 450K) were collected from a total of 1 892 colorectal samples. Pairwise differential methylation analyses between tissue types were performed for both array types to \"double evidence\" differentially methylated probes (DE DMPs). Subsequently, the identified DMPs were filtered on methylation level and used to build a binary logistic regression prediction model. Focusing on the clinically most interesting group (adenoma vs carcinoma), we identified 13 DE DMPs that could effectively discriminate between them (AUC = 0.996). We validated this model in an in-house experimental methylation dataset of 13 adenomas and 9 carcinomas. It reached a sensitivity and specificity of 96% and 95%, respectively, with an overall accuracy of 96%. Our findings raise the possibility that the 13 DE DMPs identified in this study can be used as molecular biomarkers in the clinic.</p><p><strong>Conclusions: </strong>Our analyses show that methylation biomarkers have the potential to discriminate between normal, precursor and carcinoma tissues of the colorectum. More importantly, we highlight the power of the methylome as a source of markers for discriminating between colorectal adenomas and carcinomas, which currently remains an unmet clinical need.</p>","PeriodicalId":48652,"journal":{"name":"Clinical Epigenetics","volume":"15 1","pages":"111"},"PeriodicalIF":4.4000,"publicationDate":"2023-07-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10327366/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-023-01516-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Biomarker discovery in colorectal cancer has mostly focused on methylation patterns in normal and colorectal tumor tissue, but adenomas remain understudied. Therefore, we performed the first epigenome-wide study to profile methylation of all three tissue types combined and to identify discriminatory biomarkers.
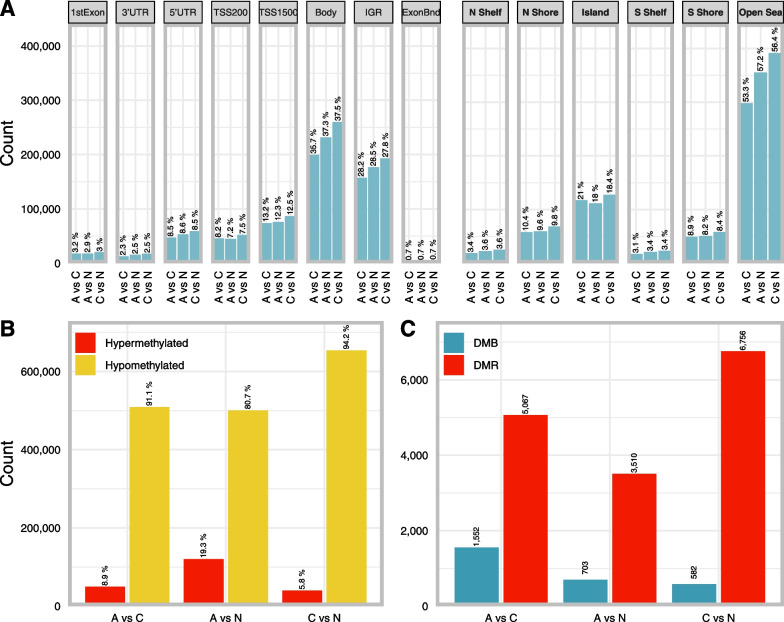
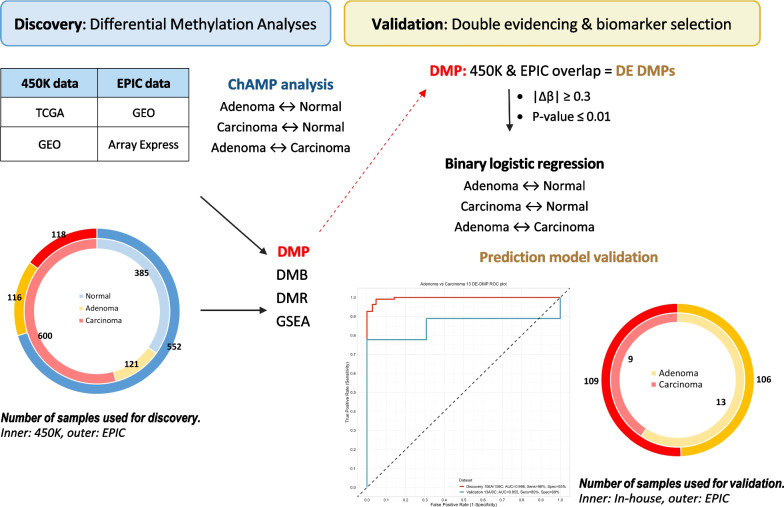
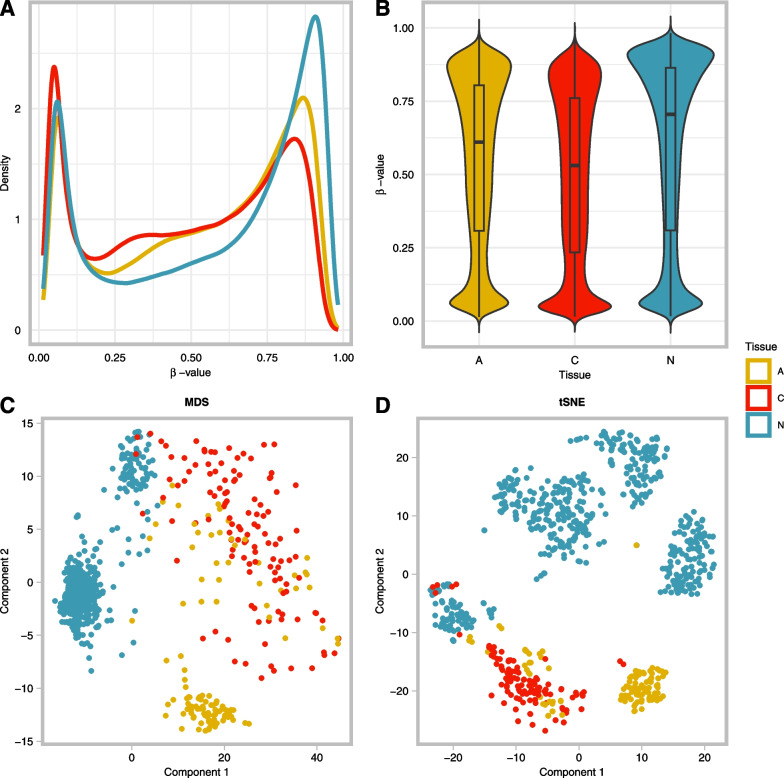
Results: Public methylation array data (Illumina EPIC and 450K) were collected from a total of 1 892 colorectal samples. Pairwise differential methylation analyses between tissue types were performed for both array types to "double evidence" differentially methylated probes (DE DMPs). Subsequently, the identified DMPs were filtered on methylation level and used to build a binary logistic regression prediction model. Focusing on the clinically most interesting group (adenoma vs carcinoma), we identified 13 DE DMPs that could effectively discriminate between them (AUC = 0.996). We validated this model in an in-house experimental methylation dataset of 13 adenomas and 9 carcinomas. It reached a sensitivity and specificity of 96% and 95%, respectively, with an overall accuracy of 96%. Our findings raise the possibility that the 13 DE DMPs identified in this study can be used as molecular biomarkers in the clinic.
Conclusions: Our analyses show that methylation biomarkers have the potential to discriminate between normal, precursor and carcinoma tissues of the colorectum. More importantly, we highlight the power of the methylome as a source of markers for discriminating between colorectal adenomas and carcinomas, which currently remains an unmet clinical need.
Clinical EpigeneticsBiochemistry, Genetics and Molecular Biology-Developmental Biology
CiteScore
8.90
自引率
5.30%
发文量
150
审稿时长
12 weeks
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
