Lucas Ferreira Teixeira, Gustavo R Krupp Prauchner, Darlan Gusso, Angela T S Wyse
{"title":"Classical Hereditary galactosemia: findings in patients and animal models.","authors":"Lucas Ferreira Teixeira, Gustavo R Krupp Prauchner, Darlan Gusso, Angela T S Wyse","doi":"10.1007/s11011-023-01281-9","DOIUrl":null,"url":null,"abstract":"<p><p>Classic galactosemia is a rare inborn error of metabolism that affects the metabolism of galactose, a sugar derived from milk and derivates. Classic galactosemia is caused by variants of the GALT gene, which lead to absent or misfolded forms of the ubiquitously present galactose-1-phosphate uridylyltransferase enzyme (GALT) driving galactose metabolites to accumulate, damaging cells from neurons to hepatocytes. The disease has different prevalence around the world due to different allele frequencies among populations and its symptoms range from cognitive and psychomotor impairment to hepatic, ophthalmological, and bone structural damage. The practice of newborn screening still varies among countries, dairy restriction treatment is a consensus despite advances in preclinical treatment strategies. Recent clinical studies in Duarte variant suggest dairy restriction could be reconsidered in these cases. Despite noteworthy advances in the classic galactosemia understanding, preclinical trials are still crucial to fully understand the pathophysiology of the disease and help propose new treatments. This review aims to report a comprehensive analysis of past studies and state of art research on galactosemia screening, its clinical and preclinical trials, and treatments with the goal of shedding light on this complex and multisystemic innate error of the metabolism.</p>","PeriodicalId":18685,"journal":{"name":"Metabolic brain disease","volume":" ","pages":"239-248"},"PeriodicalIF":3.5000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Metabolic brain disease","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11011-023-01281-9","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/13 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
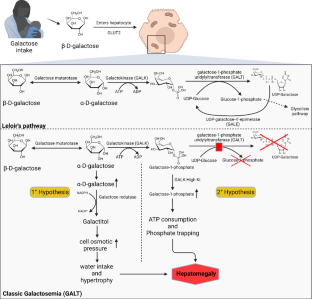
Classic galactosemia is a rare inborn error of metabolism that affects the metabolism of galactose, a sugar derived from milk and derivates. Classic galactosemia is caused by variants of the GALT gene, which lead to absent or misfolded forms of the ubiquitously present galactose-1-phosphate uridylyltransferase enzyme (GALT) driving galactose metabolites to accumulate, damaging cells from neurons to hepatocytes. The disease has different prevalence around the world due to different allele frequencies among populations and its symptoms range from cognitive and psychomotor impairment to hepatic, ophthalmological, and bone structural damage. The practice of newborn screening still varies among countries, dairy restriction treatment is a consensus despite advances in preclinical treatment strategies. Recent clinical studies in Duarte variant suggest dairy restriction could be reconsidered in these cases. Despite noteworthy advances in the classic galactosemia understanding, preclinical trials are still crucial to fully understand the pathophysiology of the disease and help propose new treatments. This review aims to report a comprehensive analysis of past studies and state of art research on galactosemia screening, its clinical and preclinical trials, and treatments with the goal of shedding light on this complex and multisystemic innate error of the metabolism.
期刊介绍:
Metabolic Brain Disease serves as a forum for the publication of outstanding basic and clinical papers on all metabolic brain disease, including both human and animal studies. The journal publishes papers on the fundamental pathogenesis of these disorders and on related experimental and clinical techniques and methodologies. Metabolic Brain Disease is directed to physicians, neuroscientists, internists, psychiatrists, neurologists, pathologists, and others involved in the research and treatment of a broad range of metabolic brain disorders.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
