Chuanyuan Wang, Shiyu Xu, Duanchen Sun, Zhi-Ping Liu
{"title":"ActivePPI: quantifying protein-protein interaction network activity with Markov random fields.","authors":"Chuanyuan Wang, Shiyu Xu, Duanchen Sun, Zhi-Ping Liu","doi":"10.1093/bioinformatics/btad567","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Protein-protein interactions (PPI) are crucial components of the biomolecular networks that enable cells to function. Biological experiments have identified a large number of PPI, and these interactions are stored in knowledge bases. However, these interactions are often restricted to specific cellular environments and conditions. Network activity can be characterized as the extent of agreement between a PPI network (PPIN) and a distinct cellular environment measured by protein mass spectrometry, and it can also be quantified as a statistical significance score. Without knowing the activity of these PPI in the cellular environments or specific phenotypes, it is impossible to reveal how these PPI perform and affect cellular functioning.</p><p><strong>Results: </strong>To calculate the activity of PPIN in different cellular conditions, we proposed a PPIN activity evaluation framework named ActivePPI to measure the consistency between network architecture and protein measurement data. ActivePPI estimates the probability density of protein mass spectrometry abundance and models PPIN using a Markov-random-field-based method. Furthermore, empirical P-value is derived based on a nonparametric permutation test to quantify the likelihood significance of the match between PPIN structure and protein abundance data. Extensive numerical experiments demonstrate the superior performance of ActivePPI and result in network activity evaluation, pathway activity assessment, and optimal network architecture tuning tasks. To summarize it succinctly, ActivePPI is a versatile tool for evaluating PPI network that can uncover the functional significance of protein interactions in crucial cellular biological processes and offer further insights into physiological phenomena.</p><p><strong>Availability and implementation: </strong>All source code and data are freely available at https://github.com/zpliulab/ActivePPI.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10516639/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad567","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Protein-protein interactions (PPI) are crucial components of the biomolecular networks that enable cells to function. Biological experiments have identified a large number of PPI, and these interactions are stored in knowledge bases. However, these interactions are often restricted to specific cellular environments and conditions. Network activity can be characterized as the extent of agreement between a PPI network (PPIN) and a distinct cellular environment measured by protein mass spectrometry, and it can also be quantified as a statistical significance score. Without knowing the activity of these PPI in the cellular environments or specific phenotypes, it is impossible to reveal how these PPI perform and affect cellular functioning.
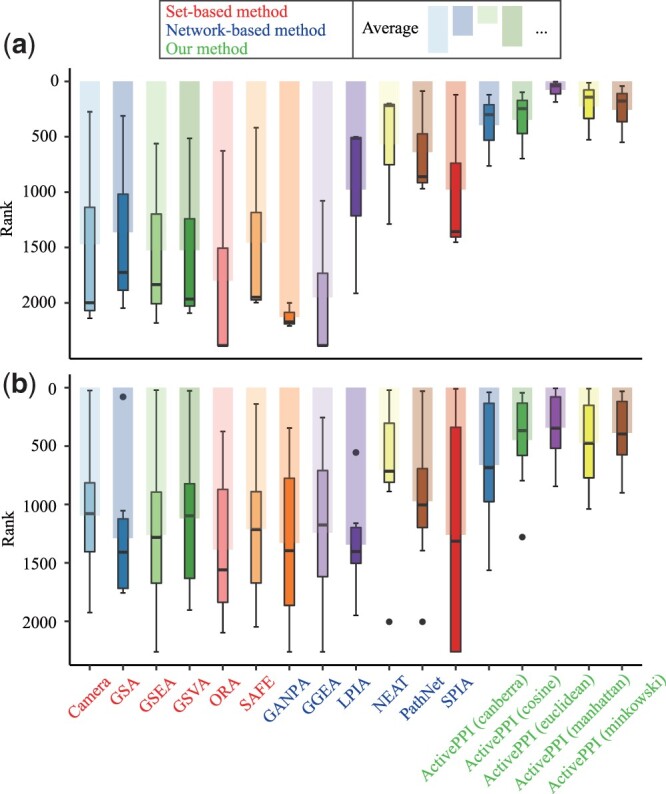
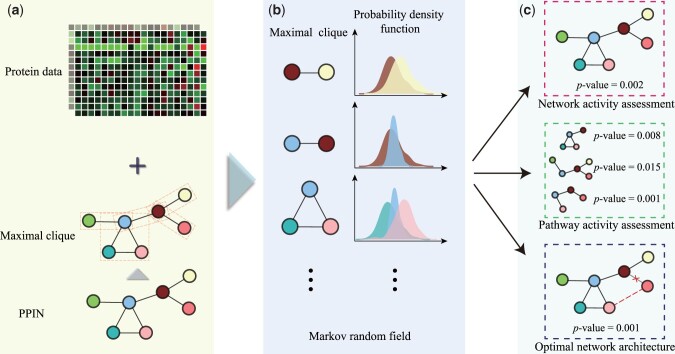
Results: To calculate the activity of PPIN in different cellular conditions, we proposed a PPIN activity evaluation framework named ActivePPI to measure the consistency between network architecture and protein measurement data. ActivePPI estimates the probability density of protein mass spectrometry abundance and models PPIN using a Markov-random-field-based method. Furthermore, empirical P-value is derived based on a nonparametric permutation test to quantify the likelihood significance of the match between PPIN structure and protein abundance data. Extensive numerical experiments demonstrate the superior performance of ActivePPI and result in network activity evaluation, pathway activity assessment, and optimal network architecture tuning tasks. To summarize it succinctly, ActivePPI is a versatile tool for evaluating PPI network that can uncover the functional significance of protein interactions in crucial cellular biological processes and offer further insights into physiological phenomena.
Availability and implementation: All source code and data are freely available at https://github.com/zpliulab/ActivePPI.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
