{"title":"Sequence-based mutation patterns at 41 Y chromosomal STRs in 2 548 father-son pairs.","authors":"Ze Liu, Guannan Long, Yubo Lang, Dahua Liu, Biao Zhang, Shaobo Yu, Fei Guo","doi":"10.1093/fsr/owad016","DOIUrl":null,"url":null,"abstract":"<p><p>A total of 2 548 unrelated healthy father-son pairs from a Northern Han Chinese population were genotyped at 41 Y chromosomal short tandem repeat (Y-STRs) including DYS19, DYS388, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS437, DYS438, DYS439, DYS444, DYS447, DYS448, DYS449, DYS456, DYS458, DYS460, DYS481, DYS518, DYS522, DYS549, DYS533, DYS557, DYS570, DYS576, DYS593, DYS596, DYS627, DYS635, DYS643, DYS645, Y-GATA-H4, DYF387S1a/b, DYF404S1a/b, DYS385a/b, and DYS527a/b. In 2 548 father samples, 2 387 unique haplotypes were detected with the haplotype diversity and discrimination capacity values of 0.999 956 608 and 0.96 741 007. The average gene diversity (GD) value was 0.6934 with a range from 0.1051 at DYS645 to 0.9657 at DYS385a/b. When comparing alleles at 24 overlapped Y-STRs between the ForenSeq™ deoxyribonucleic acid (DNA) Signature Prep Kit on the MiSeq FGx® Forensic Genomics System and the Goldeneye® DNA ID Y Plus Kit on the Applied Biosystems™ 3730 DNA Analyzer from 308 father samples in mutational pairs, 258 alleles were detected by massively parallel sequencing (MPS) typing including 156 length-based alleles that could be obtained by capillary electrophoresis (CE) typing, 95 repeat region (RR) variant alleles and seven flanking region variant alleles. Hereof, we found 16 novel RR variant alleles and firstly identified two SNPs (rs2016239814 at DYS19 and rs2089968964 at DYS448) and one 4-bp deletion (rs2053269960 at DYS439) that had been validated by the Database of Short Genetic Variation. Sanger sequencing or MPS was employed to confirm 356 mutations from 104 468 allele transfers generated from CE, where 96.63% resulted in one-step mutations, 2.25% in two-step, and 1.12% in multi-step, and the overall ratio of repeat gains <i>versus</i> losses was balanced (173 gains <i>vs.</i> 183 losses). In 308 father-son pairs, 268 pairs occurred mutations at a single locus, 33 pairs at two loci, six pairs at three loci, and one pair at four loci. The average Y-STR mutation rate at 41 Y-STRs was ⁓3.4 × 10<sup>-3</sup> (95% confidence intervals: 3.1 × 10<sup>-3</sup>-3.8 × 10<sup>-3</sup>). The mutation rates at DYS576 and DYS627 were higher than 1 × 10<sup>-2</sup> in Northern Han Chinese, whilst the mutation rates at DYF387S1a/b, DYF404S1a/b, DYS449, DYS518, and DYS570 were lower than initially defined. In this study, the classical molecular factors (the longer STR region, the more complex motif and the order father) were confirmed to drive Y-STR mutation rates increased, but the length of repeat unit did not conform to the convention. Lastly, the interactive graphical and installable StatsY was developed to facilitate forensic scientists to automatically calculate allele and haplotype frequencies, forensic parameters, and mutation rates at Y-STRs.</p><p><strong>Key points: </strong>308 of 2 548 father-son pairs from Northern Han Chinese occurred at least one mutation(s) across 41 Y-STRs.Sanger sequencing or MPS was employed to confirm those mutations generated from CE.The longer STR region, the more complex motif and the order father drove Y-STR mutation rates increased.StatsY was developed to calculate allele and haplotype frequencies, forensic parameters and mutation rates at Y-STRs.</p>","PeriodicalId":45852,"journal":{"name":"Forensic Sciences Research","volume":"8 2","pages":"152-162"},"PeriodicalIF":1.8000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10445670/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Forensic Sciences Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/fsr/owad016","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, LEGAL","Score":null,"Total":0}
引用次数: 1
Abstract
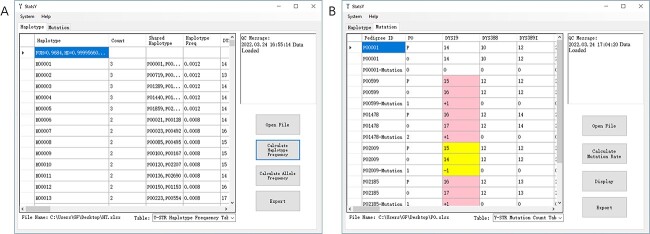
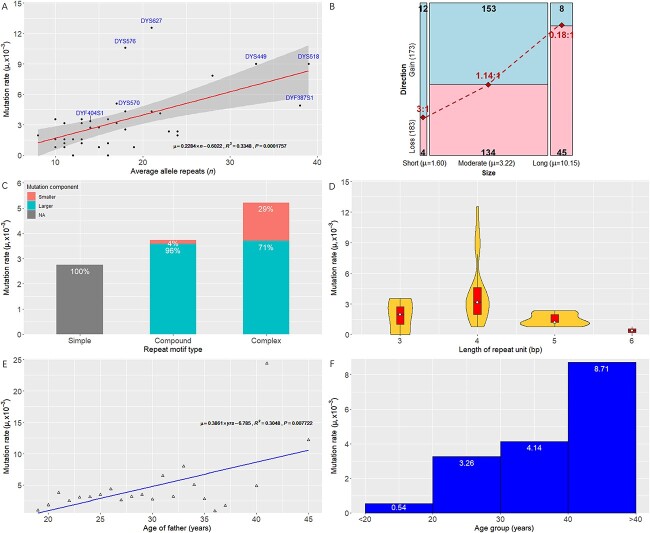
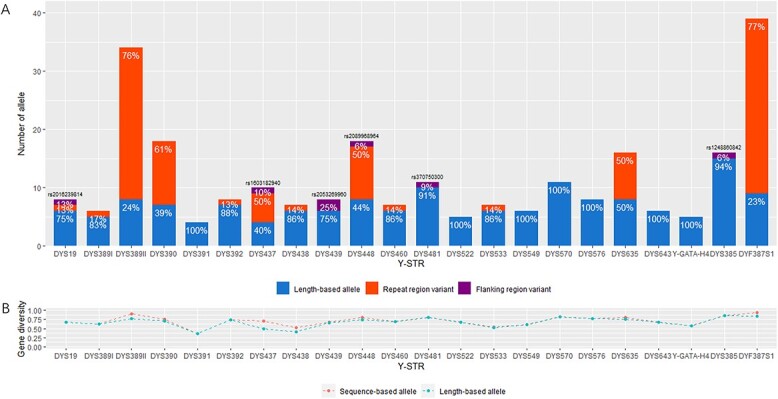
A total of 2 548 unrelated healthy father-son pairs from a Northern Han Chinese population were genotyped at 41 Y chromosomal short tandem repeat (Y-STRs) including DYS19, DYS388, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS437, DYS438, DYS439, DYS444, DYS447, DYS448, DYS449, DYS456, DYS458, DYS460, DYS481, DYS518, DYS522, DYS549, DYS533, DYS557, DYS570, DYS576, DYS593, DYS596, DYS627, DYS635, DYS643, DYS645, Y-GATA-H4, DYF387S1a/b, DYF404S1a/b, DYS385a/b, and DYS527a/b. In 2 548 father samples, 2 387 unique haplotypes were detected with the haplotype diversity and discrimination capacity values of 0.999 956 608 and 0.96 741 007. The average gene diversity (GD) value was 0.6934 with a range from 0.1051 at DYS645 to 0.9657 at DYS385a/b. When comparing alleles at 24 overlapped Y-STRs between the ForenSeq™ deoxyribonucleic acid (DNA) Signature Prep Kit on the MiSeq FGx® Forensic Genomics System and the Goldeneye® DNA ID Y Plus Kit on the Applied Biosystems™ 3730 DNA Analyzer from 308 father samples in mutational pairs, 258 alleles were detected by massively parallel sequencing (MPS) typing including 156 length-based alleles that could be obtained by capillary electrophoresis (CE) typing, 95 repeat region (RR) variant alleles and seven flanking region variant alleles. Hereof, we found 16 novel RR variant alleles and firstly identified two SNPs (rs2016239814 at DYS19 and rs2089968964 at DYS448) and one 4-bp deletion (rs2053269960 at DYS439) that had been validated by the Database of Short Genetic Variation. Sanger sequencing or MPS was employed to confirm 356 mutations from 104 468 allele transfers generated from CE, where 96.63% resulted in one-step mutations, 2.25% in two-step, and 1.12% in multi-step, and the overall ratio of repeat gains versus losses was balanced (173 gains vs. 183 losses). In 308 father-son pairs, 268 pairs occurred mutations at a single locus, 33 pairs at two loci, six pairs at three loci, and one pair at four loci. The average Y-STR mutation rate at 41 Y-STRs was ⁓3.4 × 10-3 (95% confidence intervals: 3.1 × 10-3-3.8 × 10-3). The mutation rates at DYS576 and DYS627 were higher than 1 × 10-2 in Northern Han Chinese, whilst the mutation rates at DYF387S1a/b, DYF404S1a/b, DYS449, DYS518, and DYS570 were lower than initially defined. In this study, the classical molecular factors (the longer STR region, the more complex motif and the order father) were confirmed to drive Y-STR mutation rates increased, but the length of repeat unit did not conform to the convention. Lastly, the interactive graphical and installable StatsY was developed to facilitate forensic scientists to automatically calculate allele and haplotype frequencies, forensic parameters, and mutation rates at Y-STRs.
Key points: 308 of 2 548 father-son pairs from Northern Han Chinese occurred at least one mutation(s) across 41 Y-STRs.Sanger sequencing or MPS was employed to confirm those mutations generated from CE.The longer STR region, the more complex motif and the order father drove Y-STR mutation rates increased.StatsY was developed to calculate allele and haplotype frequencies, forensic parameters and mutation rates at Y-STRs.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
