{"title":"Leber congenital amaurosis as the initial and essential manifestation in a Chinese patient with autoimmune polyglandular syndrome Type 1.","authors":"Xing Wei, Tian Zhu, Lei Wang, Ruifang Sui","doi":"10.1007/s10633-023-09953-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>Autoimmune polyglandular syndrome Type 1 (APS-1) is a rare autosomal recessive disorder caused by defects in the autoimmune regulator (AIRE) gene. Patients are generally diagnosed at ages between five and fifteen years when they exhibit three or more manifestations, most typically mucocutaneous candidiasis, autoimmune Addison's disease, and hypoparathyroidism. Our study aims to report the first case of a Chinese APS-1 patient, presented with LCA as the initial and essential clinical feature of this rare syndrome.</p><p><strong>Methods: </strong>Detailed medical and family history were recorded for the patient. Also, the comprehensive ophthalmological examinations were conducted. Whole exome sequencing (WES) was applied to screen pathogenic variants. Sanger sequencing validation and segregation analysis were further performed for confirmation.</p><p><strong>Results: </strong>A 3-year-old boy with severely impaired vision and initially referred as LCA. However, with a detailed history review, oral candidiasis, dental enamel hypoplasia, and nail candida infection were revealed. Moreover, genetic analysis revealed the homozygous c.769C>T (p.R257X) in AIRE gene (NM_000383.3) as the causative variant.</p><p><strong>Conclusion: </strong>We presented one case diagnosed with APS-1 based on clinical characteristics and genetic analysis. Our study demonstrated that LCA could serve as a warning sign for APS-1 and a potential trigger of early screening, which might prevent life-threatening complications.</p>","PeriodicalId":11207,"journal":{"name":"Documenta Ophthalmologica","volume":" ","pages":"225-232"},"PeriodicalIF":2.9000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Documenta Ophthalmologica","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10633-023-09953-8","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"OPHTHALMOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
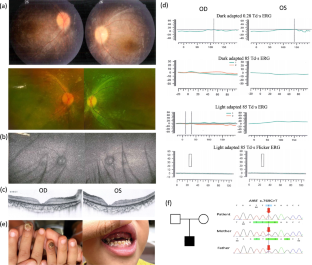
Purpose: Autoimmune polyglandular syndrome Type 1 (APS-1) is a rare autosomal recessive disorder caused by defects in the autoimmune regulator (AIRE) gene. Patients are generally diagnosed at ages between five and fifteen years when they exhibit three or more manifestations, most typically mucocutaneous candidiasis, autoimmune Addison's disease, and hypoparathyroidism. Our study aims to report the first case of a Chinese APS-1 patient, presented with LCA as the initial and essential clinical feature of this rare syndrome.
Methods: Detailed medical and family history were recorded for the patient. Also, the comprehensive ophthalmological examinations were conducted. Whole exome sequencing (WES) was applied to screen pathogenic variants. Sanger sequencing validation and segregation analysis were further performed for confirmation.
Results: A 3-year-old boy with severely impaired vision and initially referred as LCA. However, with a detailed history review, oral candidiasis, dental enamel hypoplasia, and nail candida infection were revealed. Moreover, genetic analysis revealed the homozygous c.769C>T (p.R257X) in AIRE gene (NM_000383.3) as the causative variant.
Conclusion: We presented one case diagnosed with APS-1 based on clinical characteristics and genetic analysis. Our study demonstrated that LCA could serve as a warning sign for APS-1 and a potential trigger of early screening, which might prevent life-threatening complications.
期刊介绍:
Documenta Ophthalmologica is an official publication of the International Society for Clinical Electrophysiology of Vision. The purpose of the journal is to promote the understanding and application of clinical electrophysiology of vision. Documenta Ophthalmologica will publish reviews, research articles, technical notes, brief reports and case studies which inform the readers about basic and clinical sciences related to visual electrodiagnosis and means to improve diagnosis and clinical management of patients using visual electrophysiology. Studies may involve animals or humans. In either case appropriate care must be taken to follow the Declaration of Helsinki for human subject or appropriate humane standards of animal care (e.g., the ARVO standards on Animal Care and Use).



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
