Caleb J Grohmann, Caleb M Shull, Tamar E Crum, Clint Schwab, Timothy J Safranski, Jared E Decker
{"title":"Analysis of polygenic selection in purebred and crossbred pig genomes using generation proxy selection mapping.","authors":"Caleb J Grohmann, Caleb M Shull, Tamar E Crum, Clint Schwab, Timothy J Safranski, Jared E Decker","doi":"10.1186/s12711-023-00836-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Artificial selection on quantitative traits using breeding values and selection indices in commercial livestock breeding populations causes changes in allele frequency over time at hundreds or thousands of causal loci and the surrounding genomic regions. In population genetics, this type of selection is called polygenic selection. Researchers and managers of pig breeding programs are motivated to understand the genetic basis of phenotypic diversity across genetic lines, breeds, and populations using selection mapping analyses. Here, we applied generation proxy selection mapping (GPSM), a genome-wide association analysis of single nucleotide polymorphism (SNP) genotypes (38,294-46,458 markers) of birth date, in four pig populations (15,457, 15,772, 16,595 and 8447 pigs per population) to identify loci responding to artificial selection over a period of five to ten years. Gene-drop simulation analyses were conducted to provide context for the GPSM results. Selected loci within and across each population of pigs were compared in the context of swine breeding objectives.</p><p><strong>Results: </strong>The GPSM identified 49 to 854 loci as under selection (Q-values less than 0.10) across 15 subsets of pigs based on combinations of populations. The number of significant associations increased when data were pooled across populations. In addition, several significant associations were identified in more than one population. These results indicate concurrent selection objectives, similar genetic architectures, and shared causal variants responding to selection across these pig populations. Negligible error rates (less than or equal to 0.02%) of false-positive associations were found when testing GPSM on gene-drop simulated genotypes, suggesting that GPSM distinguishes selection from random genetic drift in actual pig populations.</p><p><strong>Conclusions: </strong>This work confirms the efficacy and the negligible error rates of the GPSM method in detecting selected loci in commercial pig populations. Our results suggest shared selection objectives and genetic architectures across swine populations. The identified polygenic selection highlights loci that are important to swine production.</p>","PeriodicalId":55120,"journal":{"name":"Genetics Selection Evolution","volume":"55 1","pages":"62"},"PeriodicalIF":3.1000,"publicationDate":"2023-09-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10500877/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics Selection Evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12711-023-00836-9","RegionNum":1,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRICULTURE, DAIRY & ANIMAL SCIENCE","Score":null,"Total":0}
引用次数: 0
Abstract
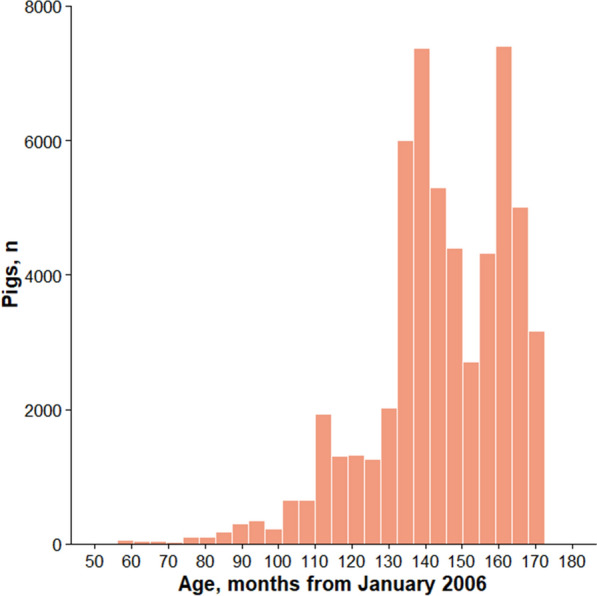
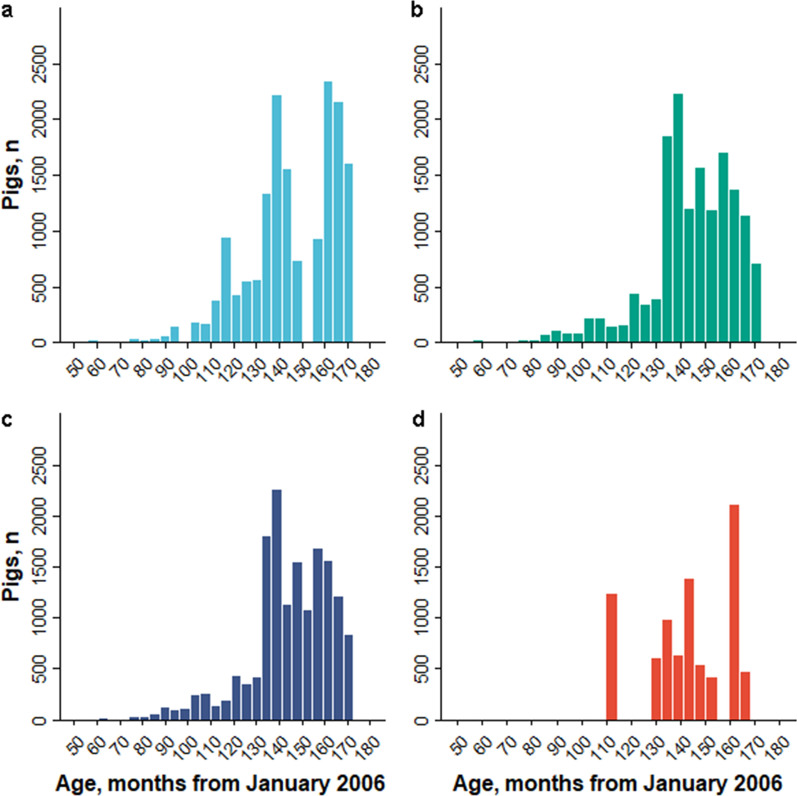
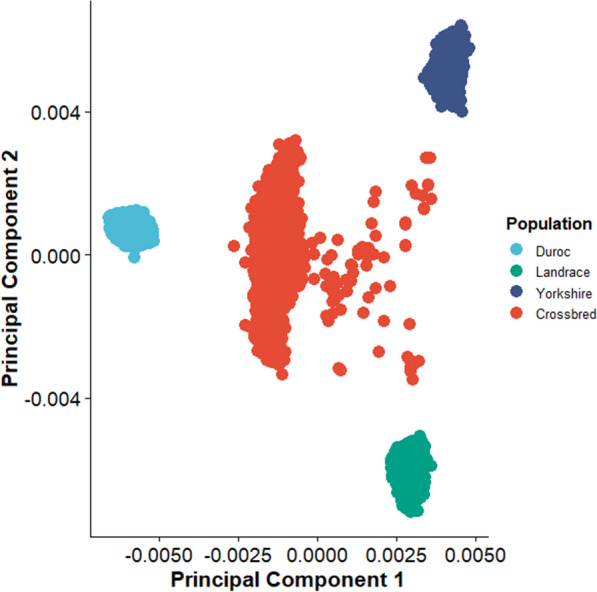
Background: Artificial selection on quantitative traits using breeding values and selection indices in commercial livestock breeding populations causes changes in allele frequency over time at hundreds or thousands of causal loci and the surrounding genomic regions. In population genetics, this type of selection is called polygenic selection. Researchers and managers of pig breeding programs are motivated to understand the genetic basis of phenotypic diversity across genetic lines, breeds, and populations using selection mapping analyses. Here, we applied generation proxy selection mapping (GPSM), a genome-wide association analysis of single nucleotide polymorphism (SNP) genotypes (38,294-46,458 markers) of birth date, in four pig populations (15,457, 15,772, 16,595 and 8447 pigs per population) to identify loci responding to artificial selection over a period of five to ten years. Gene-drop simulation analyses were conducted to provide context for the GPSM results. Selected loci within and across each population of pigs were compared in the context of swine breeding objectives.
Results: The GPSM identified 49 to 854 loci as under selection (Q-values less than 0.10) across 15 subsets of pigs based on combinations of populations. The number of significant associations increased when data were pooled across populations. In addition, several significant associations were identified in more than one population. These results indicate concurrent selection objectives, similar genetic architectures, and shared causal variants responding to selection across these pig populations. Negligible error rates (less than or equal to 0.02%) of false-positive associations were found when testing GPSM on gene-drop simulated genotypes, suggesting that GPSM distinguishes selection from random genetic drift in actual pig populations.
Conclusions: This work confirms the efficacy and the negligible error rates of the GPSM method in detecting selected loci in commercial pig populations. Our results suggest shared selection objectives and genetic architectures across swine populations. The identified polygenic selection highlights loci that are important to swine production.
期刊介绍:
Genetics Selection Evolution invites basic, applied and methodological content that will aid the current understanding and the utilization of genetic variability in domestic animal species. Although the focus is on domestic animal species, research on other species is invited if it contributes to the understanding of the use of genetic variability in domestic animals. Genetics Selection Evolution publishes results from all levels of study, from the gene to the quantitative trait, from the individual to the population, the breed or the species. Contributions concerning both the biological approach, from molecular genetics to quantitative genetics, as well as the mathematical approach, from population genetics to statistics, are welcome. Specific areas of interest include but are not limited to: gene and QTL identification, mapping and characterization, analysis of new phenotypes, high-throughput SNP data analysis, functional genomics, cytogenetics, genetic diversity of populations and breeds, genetic evaluation, applied and experimental selection, genomic selection, selection efficiency, and statistical methodology for the genetic analysis of phenotypes with quantitative and mixed inheritance.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
