Francis Banville, Dominique Gravel, Timothée Poisot
{"title":"What constrains food webs? A maximum entropy framework for predicting their structure with minimal biases.","authors":"Francis Banville, Dominique Gravel, Timothée Poisot","doi":"10.1371/journal.pcbi.1011458","DOIUrl":null,"url":null,"abstract":"<p><p>Food webs are complex ecological networks whose structure is both ecologically and statistically constrained, with many network properties being correlated with each other. Despite the recognition of these invariable relationships in food webs, the use of the principle of maximum entropy (MaxEnt) in network ecology is still rare. This is surprising considering that MaxEnt is a statistical tool precisely designed for understanding and predicting many types of constrained systems. This principle asserts that the least-biased probability distribution of a system's property, constrained by prior knowledge about that system, is the one with maximum information entropy. MaxEnt has been proven useful in many ecological modeling problems, but its application in food webs and other ecological networks is limited. Here we show how MaxEnt can be used to derive many food-web properties both analytically and heuristically. First, we show how the joint degree distribution (the joint probability distribution of the numbers of prey and predators for each species in the network) can be derived analytically using the number of species and the number of interactions in food webs. Second, we present a heuristic and flexible approach of finding a network's adjacency matrix (the network's representation in matrix format) based on simulated annealing and SVD entropy. We built two heuristic models using the connectance and the joint degree sequence as statistical constraints, respectively. We compared both models' predictions against corresponding null and neutral models commonly used in network ecology using open access data of terrestrial and aquatic food webs sampled globally (N = 257). We found that the heuristic model constrained by the joint degree sequence was a good predictor of many measures of food-web structure, especially the nestedness and motifs distribution. Specifically, our results suggest that the structure of terrestrial and aquatic food webs is mainly driven by their joint degree distribution.</p>","PeriodicalId":49688,"journal":{"name":"PLoS Computational Biology","volume":"19 9","pages":"e1011458"},"PeriodicalIF":3.6000,"publicationDate":"2023-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10503755/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Computational Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pcbi.1011458","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
Abstract
Food webs are complex ecological networks whose structure is both ecologically and statistically constrained, with many network properties being correlated with each other. Despite the recognition of these invariable relationships in food webs, the use of the principle of maximum entropy (MaxEnt) in network ecology is still rare. This is surprising considering that MaxEnt is a statistical tool precisely designed for understanding and predicting many types of constrained systems. This principle asserts that the least-biased probability distribution of a system's property, constrained by prior knowledge about that system, is the one with maximum information entropy. MaxEnt has been proven useful in many ecological modeling problems, but its application in food webs and other ecological networks is limited. Here we show how MaxEnt can be used to derive many food-web properties both analytically and heuristically. First, we show how the joint degree distribution (the joint probability distribution of the numbers of prey and predators for each species in the network) can be derived analytically using the number of species and the number of interactions in food webs. Second, we present a heuristic and flexible approach of finding a network's adjacency matrix (the network's representation in matrix format) based on simulated annealing and SVD entropy. We built two heuristic models using the connectance and the joint degree sequence as statistical constraints, respectively. We compared both models' predictions against corresponding null and neutral models commonly used in network ecology using open access data of terrestrial and aquatic food webs sampled globally (N = 257). We found that the heuristic model constrained by the joint degree sequence was a good predictor of many measures of food-web structure, especially the nestedness and motifs distribution. Specifically, our results suggest that the structure of terrestrial and aquatic food webs is mainly driven by their joint degree distribution.
期刊介绍:
PLOS Computational Biology features works of exceptional significance that further our understanding of living systems at all scales—from molecules and cells, to patient populations and ecosystems—through the application of computational methods. Readers include life and computational scientists, who can take the important findings presented here to the next level of discovery.
Research articles must be declared as belonging to a relevant section. More information about the sections can be found in the submission guidelines.
Research articles should model aspects of biological systems, demonstrate both methodological and scientific novelty, and provide profound new biological insights.
Generally, reliability and significance of biological discovery through computation should be validated and enriched by experimental studies. Inclusion of experimental validation is not required for publication, but should be referenced where possible. Inclusion of experimental validation of a modest biological discovery through computation does not render a manuscript suitable for PLOS Computational Biology.
Research articles specifically designated as Methods papers should describe outstanding methods of exceptional importance that have been shown, or have the promise to provide new biological insights. The method must already be widely adopted, or have the promise of wide adoption by a broad community of users. Enhancements to existing published methods will only be considered if those enhancements bring exceptional new capabilities.
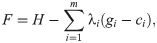
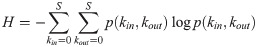
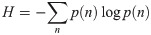



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
