{"title":"MuTATE-an R package for comprehensive multi-objective molecular modeling.","authors":"Sarah G Ayton, Víctor Treviño","doi":"10.1093/bioinformatics/btad507","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Comprehensive multi-omics studies have driven advances in disease modeling for effective precision medicine but pose a challenge for existing machine-learning approaches, which have limited interpretability across clinical endpoints. Automated, comprehensive disease modeling requires a machine-learning approach that can simultaneously identify disease subgroups and their defining molecular biomarkers by explaining multiple clinical endpoints. Current tools are restricted to individual endpoints or limited variable types, necessitate advanced computation skills, and require resource-intensive manual expert interpretation.</p><p><strong>Results: </strong>We developed Multi-Target Automated Tree Engine (MuTATE) for automated and comprehensive molecular modeling, which enables user-friendly multi-objective decision tree construction and visualization of relationships between molecular biomarkers and patient subgroups characterized by multiple clinical endpoints. MuTATE incorporates multiple targets throughout model construction and allows for target weights, enabling construction of interpretable decision trees that provide insights into disease heterogeneity and molecular signatures. MuTATE eliminates the need for manual synthesis of multiple non-explainable models, making it highly efficient and accessible for bioinformaticians and clinicians. The flexibility and versatility of MuTATE make it applicable to a wide range of complex diseases, including cancer, where it can improve therapeutic decisions by providing comprehensive molecular insights for precision medicine. MuTATE has the potential to transform biomarker discovery and subtype identification, leading to more effective and personalized treatment strategies in precision medicine, and advancing our understanding of disease mechanisms at the molecular level.</p><p><strong>Availability and implementation: </strong>MuTATE is freely available at GitHub (https://github.com/SarahAyton/MuTATE) under the GPLv3 license.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10500092/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad507","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Comprehensive multi-omics studies have driven advances in disease modeling for effective precision medicine but pose a challenge for existing machine-learning approaches, which have limited interpretability across clinical endpoints. Automated, comprehensive disease modeling requires a machine-learning approach that can simultaneously identify disease subgroups and their defining molecular biomarkers by explaining multiple clinical endpoints. Current tools are restricted to individual endpoints or limited variable types, necessitate advanced computation skills, and require resource-intensive manual expert interpretation.
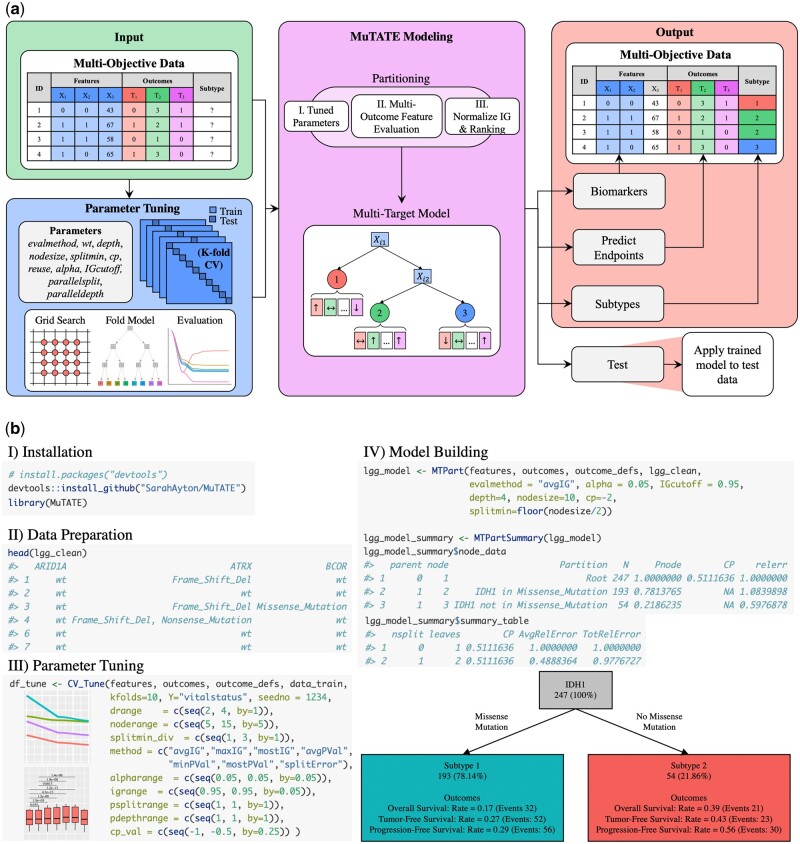
Results: We developed Multi-Target Automated Tree Engine (MuTATE) for automated and comprehensive molecular modeling, which enables user-friendly multi-objective decision tree construction and visualization of relationships between molecular biomarkers and patient subgroups characterized by multiple clinical endpoints. MuTATE incorporates multiple targets throughout model construction and allows for target weights, enabling construction of interpretable decision trees that provide insights into disease heterogeneity and molecular signatures. MuTATE eliminates the need for manual synthesis of multiple non-explainable models, making it highly efficient and accessible for bioinformaticians and clinicians. The flexibility and versatility of MuTATE make it applicable to a wide range of complex diseases, including cancer, where it can improve therapeutic decisions by providing comprehensive molecular insights for precision medicine. MuTATE has the potential to transform biomarker discovery and subtype identification, leading to more effective and personalized treatment strategies in precision medicine, and advancing our understanding of disease mechanisms at the molecular level.
Availability and implementation: MuTATE is freely available at GitHub (https://github.com/SarahAyton/MuTATE) under the GPLv3 license.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
