{"title":"Genetic risk score for ovarian cancer based on chromosomal-scale length variation.","authors":"Christopher Toh, James P Brody","doi":"10.1186/s13040-021-00253-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Twin studies indicate that a substantial fraction of ovarian cancers should be predictable from genetic testing. Genetic risk scores can stratify women into different classes of risk. Higher risk women can be treated or screened for ovarian cancer, which should reduce ovarian cancer death rates. However, current ovarian cancer genetic risk scores do not work that well. We developed a genetic risk score based on variations in the length of chromosomes.</p><p><strong>Methods: </strong>We evaluated this genetic risk score using data collected by The Cancer Genome Atlas. We synthesized a dataset of 414 women who had ovarian serous carcinoma and 4225 women who had no form of ovarian cancer. We characterized each woman by 22 numbers, representing the length of each chromosome in their germ line DNA. We used a gradient boosting machine to build a classifier that can predict whether a woman had been diagnosed with ovarian cancer.</p><p><strong>Results: </strong>The genetic risk score based on chromosomal-scale length variation could stratify women such that the highest 20% had a 160x risk (95% confidence interval 50x-450x) compared to the lowest 20%. The genetic risk score we developed had an area under the curve of the receiver operating characteristic curve of 0.88 (95% confidence interval 0.86-0.91).</p><p><strong>Conclusion: </strong>A genetic risk score based on chromosomal-scale length variation of germ line DNA provides an effective means of predicting whether or not a woman will develop ovarian cancer.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"14 1","pages":"18"},"PeriodicalIF":6.1000,"publicationDate":"2021-03-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13040-021-00253-y","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-021-00253-y","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 5
Abstract
Introduction: Twin studies indicate that a substantial fraction of ovarian cancers should be predictable from genetic testing. Genetic risk scores can stratify women into different classes of risk. Higher risk women can be treated or screened for ovarian cancer, which should reduce ovarian cancer death rates. However, current ovarian cancer genetic risk scores do not work that well. We developed a genetic risk score based on variations in the length of chromosomes.
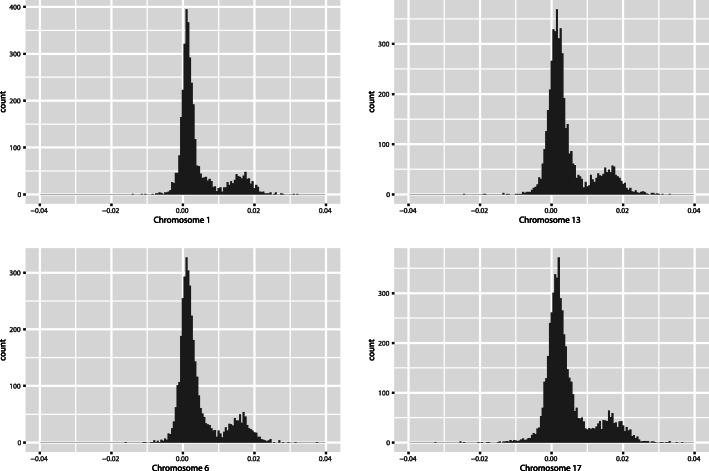
Methods: We evaluated this genetic risk score using data collected by The Cancer Genome Atlas. We synthesized a dataset of 414 women who had ovarian serous carcinoma and 4225 women who had no form of ovarian cancer. We characterized each woman by 22 numbers, representing the length of each chromosome in their germ line DNA. We used a gradient boosting machine to build a classifier that can predict whether a woman had been diagnosed with ovarian cancer.
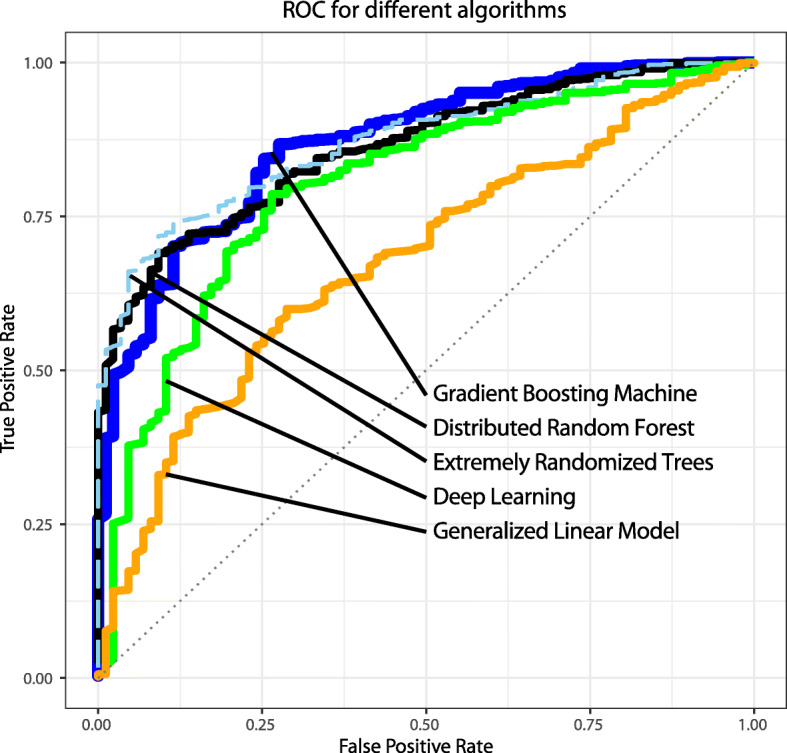
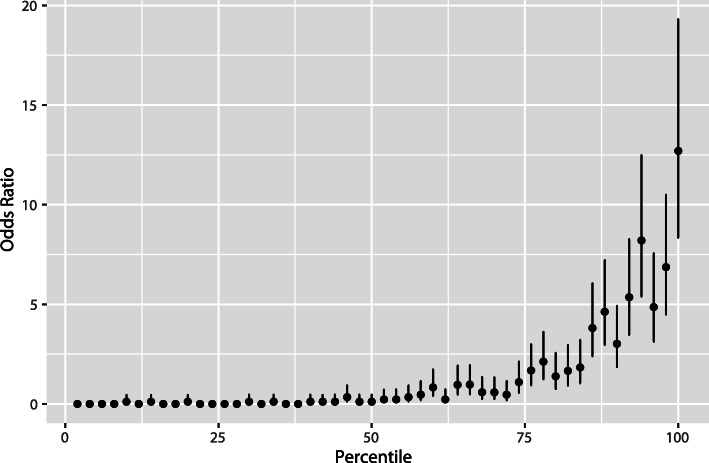
Results: The genetic risk score based on chromosomal-scale length variation could stratify women such that the highest 20% had a 160x risk (95% confidence interval 50x-450x) compared to the lowest 20%. The genetic risk score we developed had an area under the curve of the receiver operating characteristic curve of 0.88 (95% confidence interval 0.86-0.91).
Conclusion: A genetic risk score based on chromosomal-scale length variation of germ line DNA provides an effective means of predicting whether or not a woman will develop ovarian cancer.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
