{"title":"Improving the accuracy of GIPAW chemical shielding calculations with cluster and fragment corrections","authors":"Joshua D. Hartman , James K. Harper","doi":"10.1016/j.ssnmr.2022.101832","DOIUrl":null,"url":null,"abstract":"<div><p><span>Ab initio methods<span> for predicting NMR parameters in the solid state are an essential tool for assigning experimental spectra and play an increasingly important role in structural characterizations. Recently, a molecular correction (MC) technique has been developed which combines the strengths<span> of plane-wave methods (GIPAW) with single molecule calculations employing Gaussian basis sets. The GIPAW + MC method relies on a periodic calculation performed at a lower level of theory to model the crystalline environment. The GIPAW result is then corrected using a single molecule calculation performed at a higher level of theory. The success of the GIPAW + MC method in predicting a range of NMR parameters is a result of the highly local character of the tensors underlying the NMR observable. However, in applications involving strong intermolecular interactions we find that expanding the region treated at the higher level of theory more accurately captures local many-body contributions to the </span></span></span><span><math><mmultiscripts><mrow><mi>N</mi></mrow><none></none><none></none><mprescripts></mprescripts><none></none><mrow><mn>15</mn></mrow></mmultiscripts></math></span> NMR chemical shielding (CS) tensor. We propose alternative corrections to GIPAW which capture interactions between adjacent molecules at a higher level of theory using either fragment or cluster-based calculations. Benchmark calculations performed on <span><math><mmultiscripts><mrow><mi>N</mi></mrow><none></none><none></none><mprescripts></mprescripts><none></none><mrow><mn>15</mn></mrow></mmultiscripts></math></span> and <span><math><mmultiscripts><mrow><mi>C</mi></mrow><none></none><none></none><mprescripts></mprescripts><none></none><mrow><mn>13</mn></mrow></mmultiscripts></math></span> data sets show that these advanced GIPAW-corrected calculations improve the accuracy of chemical shielding tensor predictions relative to existing methods. Specifically, cluster-based <span><math><mmultiscripts><mrow><mi>N</mi></mrow><none></none><none></none><mprescripts></mprescripts><none></none><mrow><mn>15</mn></mrow></mmultiscripts></math></span> corrections show a 24% and 17% reduction in RMS error relative to GIPAW and GIPAW + MC calculations, respectively. Comparing the benchmark data sets using multiple computational models demonstrates that <span><math><mmultiscripts><mrow><mi>N</mi></mrow><none></none><none></none><mprescripts></mprescripts><none></none><mrow><mn>15</mn></mrow></mmultiscripts></math></span> CS tensor calculations are significantly more sensitive to intermolecular interactions relative to <span><math><mmultiscripts><mrow><mi>C</mi></mrow><none></none><none></none><mprescripts></mprescripts><none></none><mrow><mn>13</mn></mrow></mmultiscripts></math></span><span>. However, fragment and cluster-based corrections that include direct hydrogen bond<span> partners are sufficient for optimizing the accuracy of GIPAW-corrected methods. Finally, GIPAW-corrected methods are applied to the particularly challenging NMR spectral assignment of guanosine<span> dihydrate which contains two guanosine molecules in the asymmetric unit.</span></span></span></p></div>","PeriodicalId":21937,"journal":{"name":"Solid state nuclear magnetic resonance","volume":"122 ","pages":"Article 101832"},"PeriodicalIF":2.4000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Solid state nuclear magnetic resonance","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0926204022000613","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/9/24 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 3
Abstract
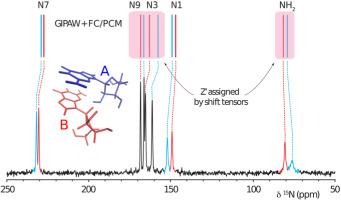
Ab initio methods for predicting NMR parameters in the solid state are an essential tool for assigning experimental spectra and play an increasingly important role in structural characterizations. Recently, a molecular correction (MC) technique has been developed which combines the strengths of plane-wave methods (GIPAW) with single molecule calculations employing Gaussian basis sets. The GIPAW + MC method relies on a periodic calculation performed at a lower level of theory to model the crystalline environment. The GIPAW result is then corrected using a single molecule calculation performed at a higher level of theory. The success of the GIPAW + MC method in predicting a range of NMR parameters is a result of the highly local character of the tensors underlying the NMR observable. However, in applications involving strong intermolecular interactions we find that expanding the region treated at the higher level of theory more accurately captures local many-body contributions to the NMR chemical shielding (CS) tensor. We propose alternative corrections to GIPAW which capture interactions between adjacent molecules at a higher level of theory using either fragment or cluster-based calculations. Benchmark calculations performed on and data sets show that these advanced GIPAW-corrected calculations improve the accuracy of chemical shielding tensor predictions relative to existing methods. Specifically, cluster-based corrections show a 24% and 17% reduction in RMS error relative to GIPAW and GIPAW + MC calculations, respectively. Comparing the benchmark data sets using multiple computational models demonstrates that CS tensor calculations are significantly more sensitive to intermolecular interactions relative to . However, fragment and cluster-based corrections that include direct hydrogen bond partners are sufficient for optimizing the accuracy of GIPAW-corrected methods. Finally, GIPAW-corrected methods are applied to the particularly challenging NMR spectral assignment of guanosine dihydrate which contains two guanosine molecules in the asymmetric unit.
期刊介绍:
The journal Solid State Nuclear Magnetic Resonance publishes original manuscripts of high scientific quality dealing with all experimental and theoretical aspects of solid state NMR. This includes advances in instrumentation, development of new experimental techniques and methodology, new theoretical insights, new data processing and simulation methods, and original applications of established or novel methods to scientific problems.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
