Pavel Flegontov, Ulaş Işıldak, Robert Maier, Eren Yüncü, Piya Changmai, David Reich
{"title":"Modeling of African population history using f-statistics is biased when applying all previously proposed SNP ascertainment schemes.","authors":"Pavel Flegontov, Ulaş Işıldak, Robert Maier, Eren Yüncü, Piya Changmai, David Reich","doi":"10.1371/journal.pgen.1010931","DOIUrl":null,"url":null,"abstract":"<p><p>f-statistics have emerged as a first line of analysis for making inferences about demographic history from genome-wide data. Not only are they guaranteed to allow robust tests of the fits of proposed models of population history to data when analyzing full genome sequencing data-that is, all single nucleotide polymorphisms (SNPs) in the individuals being analyzed-but they are also guaranteed to allow robust tests of models for SNPs ascertained as polymorphic in a population that is an outgroup in a phylogenetic sense to all groups being analyzed. True \"outgroup ascertainment\" is in practice impossible in humans because our species has arisen from a substructured ancestral population that does not descend from a homogeneous ancestral population going back many hundreds of thousands of years into the past. However, initial studies suggested that non-outgroup-ascertainment schemes might produce robust enough results using f-statistics, and that motivated widespread fitting of models to data using non-outgroup-ascertained SNP panels such as the \"Affymetrix Human Origins array\" which has been genotyped on thousands of modern individuals from hundreds of populations, or the \"1240k\" in-solution enrichment reagent which has been the source of about 70% of published genome-wide data for ancient humans. In this study, we show that while analyses of population history using such panels work well for studies of relationships among non-African populations and one African outgroup, when co-modeling more than one sub-Saharan African and/or archaic human groups (Neanderthals and Denisovans), fitting of f-statistics to such SNP sets is expected to frequently lead to false rejection of true demographic histories, and failure to reject incorrect models. Analyzing panels of SNPs polymorphic in archaic humans, which has been suggested as a solution for the ascertainment problem, has limited statistical power and retains important biases. However, by carrying out simulations of diverse demographic histories, we show that bias in inferences based on f-statistics can be minimized by ascertaining on variants common in a union of diverse African groups; such ascertainment retains high statistical power while allowing co-analysis of archaic and modern groups.</p>","PeriodicalId":20266,"journal":{"name":"PLoS Genetics","volume":"19 9","pages":"e1010931"},"PeriodicalIF":3.7000,"publicationDate":"2023-09-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10508636/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pgen.1010931","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
Abstract
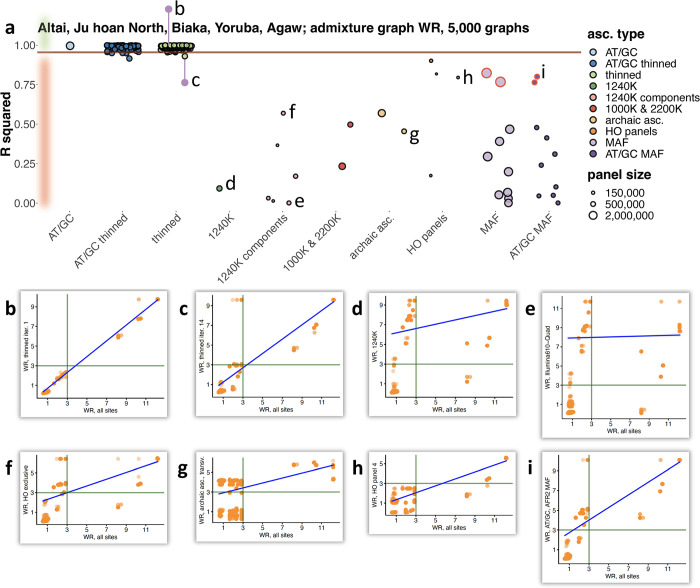
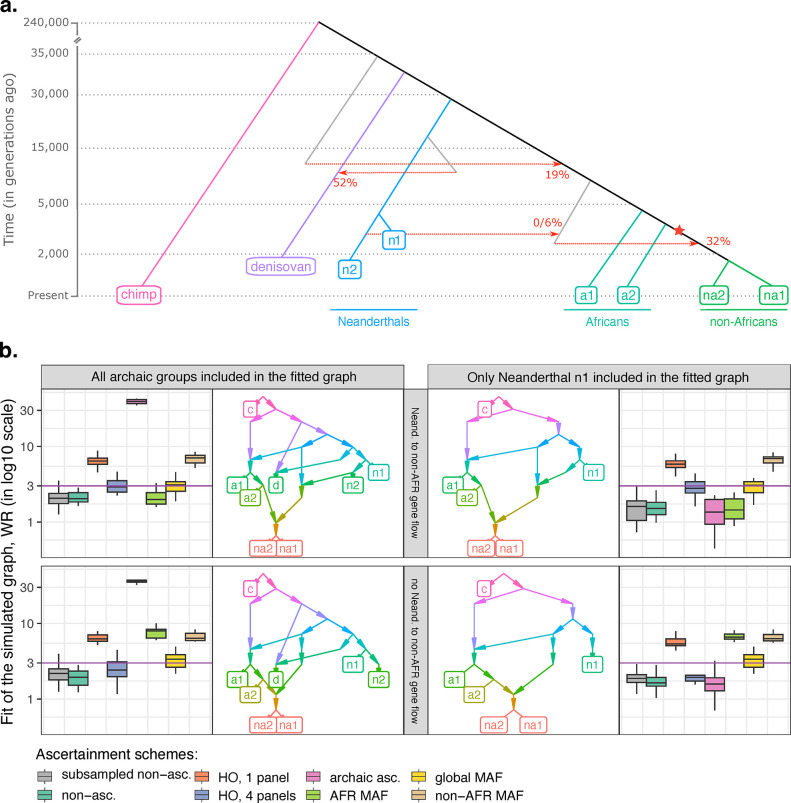
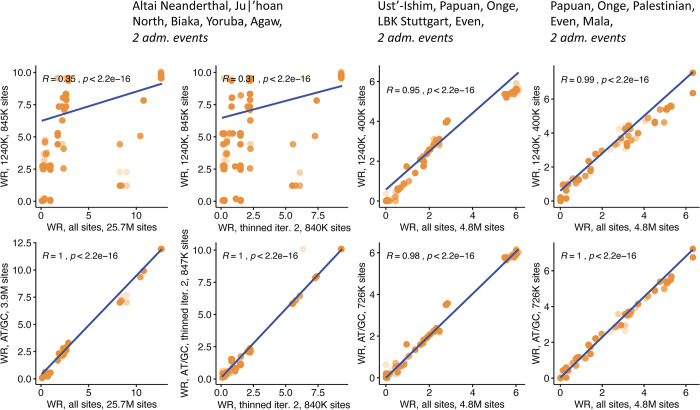
f-statistics have emerged as a first line of analysis for making inferences about demographic history from genome-wide data. Not only are they guaranteed to allow robust tests of the fits of proposed models of population history to data when analyzing full genome sequencing data-that is, all single nucleotide polymorphisms (SNPs) in the individuals being analyzed-but they are also guaranteed to allow robust tests of models for SNPs ascertained as polymorphic in a population that is an outgroup in a phylogenetic sense to all groups being analyzed. True "outgroup ascertainment" is in practice impossible in humans because our species has arisen from a substructured ancestral population that does not descend from a homogeneous ancestral population going back many hundreds of thousands of years into the past. However, initial studies suggested that non-outgroup-ascertainment schemes might produce robust enough results using f-statistics, and that motivated widespread fitting of models to data using non-outgroup-ascertained SNP panels such as the "Affymetrix Human Origins array" which has been genotyped on thousands of modern individuals from hundreds of populations, or the "1240k" in-solution enrichment reagent which has been the source of about 70% of published genome-wide data for ancient humans. In this study, we show that while analyses of population history using such panels work well for studies of relationships among non-African populations and one African outgroup, when co-modeling more than one sub-Saharan African and/or archaic human groups (Neanderthals and Denisovans), fitting of f-statistics to such SNP sets is expected to frequently lead to false rejection of true demographic histories, and failure to reject incorrect models. Analyzing panels of SNPs polymorphic in archaic humans, which has been suggested as a solution for the ascertainment problem, has limited statistical power and retains important biases. However, by carrying out simulations of diverse demographic histories, we show that bias in inferences based on f-statistics can be minimized by ascertaining on variants common in a union of diverse African groups; such ascertainment retains high statistical power while allowing co-analysis of archaic and modern groups.
期刊介绍:
PLOS Genetics is run by an international Editorial Board, headed by the Editors-in-Chief, Greg Barsh (HudsonAlpha Institute of Biotechnology, and Stanford University School of Medicine) and Greg Copenhaver (The University of North Carolina at Chapel Hill).
Articles published in PLOS Genetics are archived in PubMed Central and cited in PubMed.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
