{"title":"Study of trioleoylglycerol two-layer and adiposome cross-section mimicking four-layer systems through atomic-level simulations.","authors":"Ahmed Hammad Mirza","doi":"10.1063/4.0000168","DOIUrl":null,"url":null,"abstract":"<p><p>Adiposomes are artificially prepared lipid droplet (LD)-mimetic structures, which, unlike LDs, do not harbor proteins. The dynamics of interaction between triacylglycerols (TAGs), drug molecule, and phospholipids in adiposomes is currently not well-established. Trioleoylglycerol (TOG) molecule was divided into three parts: two oleoyl tails and one 2-monooleoylglycerol (MOG). Forcefield parameters for two oleoyl tails were adopted from the AMBER18 repository while that of the MOG forcefield was taken from the literature. Charge correction was performed on the MOG forcefield before its utilization. After charge correction, the resulting TOG molecule had zero charge. TOG bilayer (2L) and tetralayer (4L) systems were prepared and simulated. TOG bilayer (2L) systems-modeled from two different initial conformations, the TOG3 conformation and the TOG2:1 conformation-showed that TOG2:1 conformation was more prevailing irrespective of the starting conformation and was subsequently used in further simulations. The hydrated TOG 2L system showed TOG-water solution solubility of 0.051 mol L<sup>-1</sup> which is near experimental values. This validated the correct parameterization of the TOG molecule. The simulations of 4L systems showed stable membrane behaviors toward the end of simulations. It was also observed that in the 4L system, the TOG molecules showed the formation of micelles with the drug molecule. Almost six TOGs remained continuously in contact with the drug molecule throughout the simulation. The availability of charge-corrected TOG parameterization is expected to equip future studies with a framework for molecular dynamics simulations of adiposomes and/or LDs at the atomic level.</p>","PeriodicalId":74877,"journal":{"name":"","volume":"9 6","pages":"064701"},"PeriodicalIF":0.0,"publicationDate":"2022-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9726221/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"","FirstCategoryId":"101","ListUrlMain":"https://doi.org/10.1063/4.0000168","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/11/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
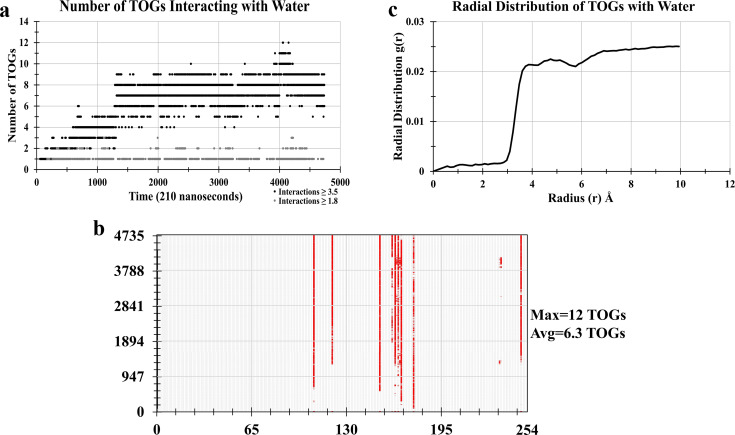
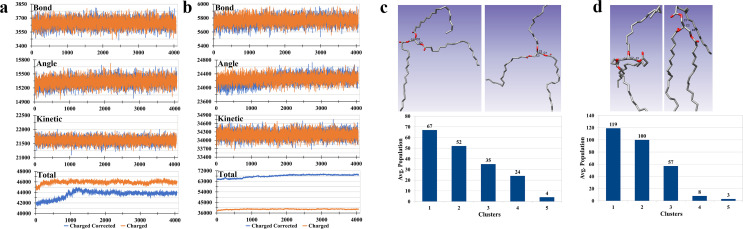
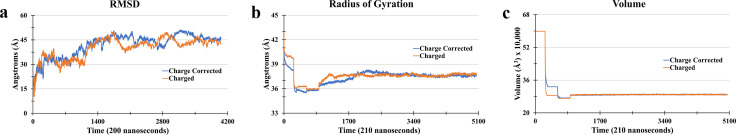
Adiposomes are artificially prepared lipid droplet (LD)-mimetic structures, which, unlike LDs, do not harbor proteins. The dynamics of interaction between triacylglycerols (TAGs), drug molecule, and phospholipids in adiposomes is currently not well-established. Trioleoylglycerol (TOG) molecule was divided into three parts: two oleoyl tails and one 2-monooleoylglycerol (MOG). Forcefield parameters for two oleoyl tails were adopted from the AMBER18 repository while that of the MOG forcefield was taken from the literature. Charge correction was performed on the MOG forcefield before its utilization. After charge correction, the resulting TOG molecule had zero charge. TOG bilayer (2L) and tetralayer (4L) systems were prepared and simulated. TOG bilayer (2L) systems-modeled from two different initial conformations, the TOG3 conformation and the TOG2:1 conformation-showed that TOG2:1 conformation was more prevailing irrespective of the starting conformation and was subsequently used in further simulations. The hydrated TOG 2L system showed TOG-water solution solubility of 0.051 mol L-1 which is near experimental values. This validated the correct parameterization of the TOG molecule. The simulations of 4L systems showed stable membrane behaviors toward the end of simulations. It was also observed that in the 4L system, the TOG molecules showed the formation of micelles with the drug molecule. Almost six TOGs remained continuously in contact with the drug molecule throughout the simulation. The availability of charge-corrected TOG parameterization is expected to equip future studies with a framework for molecular dynamics simulations of adiposomes and/or LDs at the atomic level.
脂肪体是人工制备的仿脂滴(LD)结构,与 LD 不同,它不含有蛋白质。脂肪体中的三酰甘油(TAG)、药物分子和磷脂之间的相互作用动力学目前尚未得到很好的证实。三油酰甘油(TOG)分子被分为三部分:两个油酰尾和一个 2-单油酰甘油(MOG)。两个油酰基尾部的力场参数来自 AMBER18 数据库,而 MOG 力场的参数则来自文献。在使用 MOG 力场之前对其进行了电荷校正。经过电荷校正后,得到的 TOG 分子电荷为零。制备并模拟了 TOG 双层(2L)和四层(4L)系统。从两种不同的初始构象(TOG3构象和TOG2:1构象)模拟的TOG双分子层(2L)系统显示,无论初始构象如何,TOG2:1构象都更为普遍,因此被用于进一步的模拟。水合 TOG 2L 系统显示 TOG 水溶液溶解度为 0.051 mol L-1,接近实验值。这验证了对 TOG 分子参数化的正确性。4L 系统的模拟结果表明,在模拟的最后阶段,膜的行为比较稳定。还观察到在 4L 系统中,TOG 分子与药物分子形成胶束。在整个模拟过程中,近六个 TOG 始终与药物分子保持接触。电荷校正 TOG 参数化的可用性有望为今后的研究提供原子水平的脂肪体和/或 LD 的分子动力学模拟框架。


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
