Raquel Baviera-Muñoz, Lidón Carretero-Vilarroig, Juan Francisco Vázquez-Costa, Carlos Morata-Martínez, Marina Campins-Romeu, Nuria Muelas, Isabel Sastre-Bataller, Irene Martínez-Torres, Julia Pérez-García, Rafael Sivera, Teresa Sevilla, Juan J Vilchez, Teresa Jaijo, Carmen Espinós, Jose M Millán, Luis Bataller, Elena Aller
{"title":"Diagnostic Efficacy of Genetic Studies in a Series of Hereditary Cerebellar Ataxias in Eastern Spain.","authors":"Raquel Baviera-Muñoz, Lidón Carretero-Vilarroig, Juan Francisco Vázquez-Costa, Carlos Morata-Martínez, Marina Campins-Romeu, Nuria Muelas, Isabel Sastre-Bataller, Irene Martínez-Torres, Julia Pérez-García, Rafael Sivera, Teresa Sevilla, Juan J Vilchez, Teresa Jaijo, Carmen Espinós, Jose M Millán, Luis Bataller, Elena Aller","doi":"10.1212/NXG.0000000000200038","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>To determine the diagnostic efficacy of clinical exome-targeted sequencing (CES) and spinocerebellar ataxia 36 (SCA36) screening in a real-life cohort of patients with cerebellar ataxia (CA) from Eastern Spain.</p><p><strong>Methods: </strong>A total of 130 unrelated patients with CA, negative for common trinucleotide repeat expansions (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, dentatorubral pallidoluysian atrophy [DRPLA], and Friedreich ataxia), were studied with CES. Bioinformatic and genotype-phenotype analyses were performed to assess the pathogenicity of the variants encountered. Copy number variants were analyzed when appropriate. In undiagnosed dominant and sporadic cases, repeat primed PCR was used to screen for the presence of a repeat expansion in the <i>NOP56</i> gene.</p><p><strong>Results: </strong>CES identified pathogenic or likely pathogenic variants in 50 families (39%), including 23 novel variants. Overall, there was a high genetic heterogeneity, and the most frequent genetic diagnosis was <i>SPG7</i> (n = 15), followed by <i>SETX</i> (n = 6), <i>CACNA1A</i> (n = 5), <i>POLR3A</i> (n = 4), and <i>SYNE1</i> (n = 3). In addition, 17 families displayed likely pathogenic/pathogenic variants in 14 different genes: <i>KCND3</i> (n = 2), <i>KIF1C</i> (n = 2), <i>CYP27A1A</i> (n = 2), <i>AFG3L2</i> (n = 1), <i>ANO10</i> (n = 1), <i>CAPN1</i> (n = 1), <i>CWF19L1</i> (n = 1), <i>ITPR1</i> (n = 1), <i>KCNA1</i> (n = 1), <i>OPA1</i> (n = 1), <i>PNPLA6</i> (n = 1), <i>SPG11</i> (n = 1), <i>SPTBN2</i> (n = 1), and <i>TPP1</i> (n = 1). Twenty-two novel variants were characterized. SCA36 was diagnosed in 11 families, all with autosomal dominant (AD) presentation. SCA36 screening increased the total diagnostic rate to 47% (n = 61/130). Ultimately, undiagnosed patients showed delayed age at onset (<i>p</i> < 0.05) and were more frequently sporadic.</p><p><strong>Discussion: </strong>Our study provides insight into the genetic landscape of CA in Eastern Spain. Although CES was an effective approach to capture genetic heterogeneity, most patients remained undiagnosed. SCA36 was found to be a relatively frequent form and, therefore, should be tested prior to CES in familial AD presentations in particular geographical regions.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"8 6","pages":"e200038"},"PeriodicalIF":3.7000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6d/fd/NXG-2022-200041.PMC9749935.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200038","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
Background and objectives: To determine the diagnostic efficacy of clinical exome-targeted sequencing (CES) and spinocerebellar ataxia 36 (SCA36) screening in a real-life cohort of patients with cerebellar ataxia (CA) from Eastern Spain.
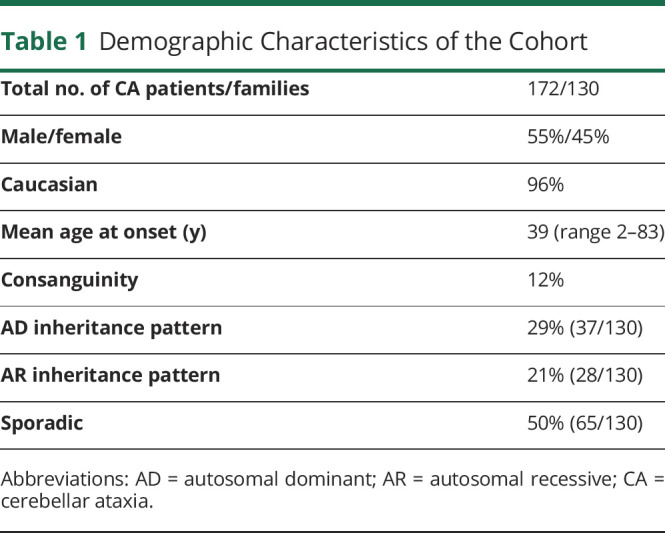
Methods: A total of 130 unrelated patients with CA, negative for common trinucleotide repeat expansions (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, dentatorubral pallidoluysian atrophy [DRPLA], and Friedreich ataxia), were studied with CES. Bioinformatic and genotype-phenotype analyses were performed to assess the pathogenicity of the variants encountered. Copy number variants were analyzed when appropriate. In undiagnosed dominant and sporadic cases, repeat primed PCR was used to screen for the presence of a repeat expansion in the NOP56 gene.
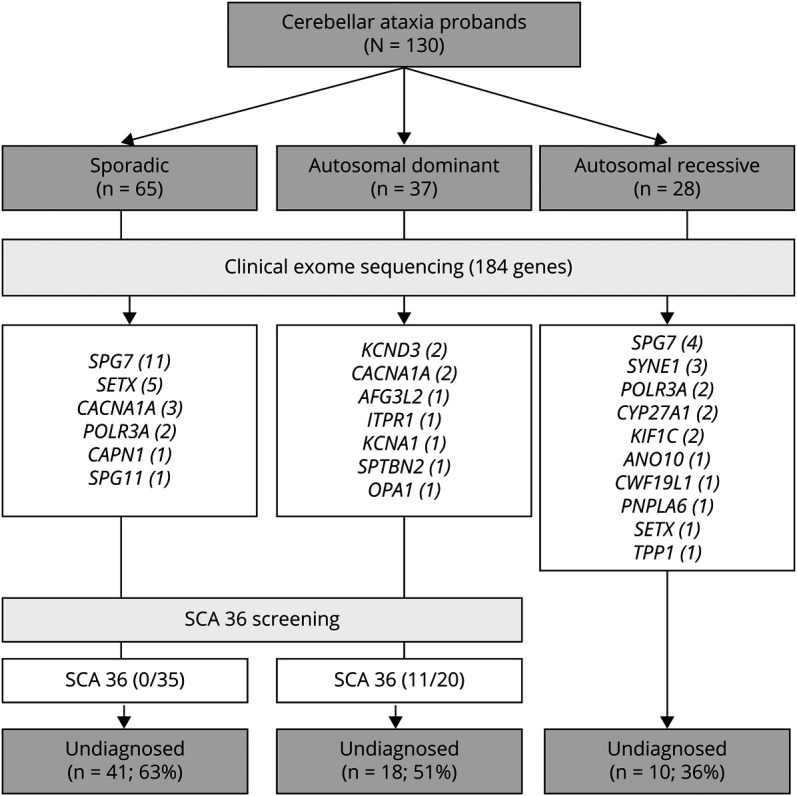
Results: CES identified pathogenic or likely pathogenic variants in 50 families (39%), including 23 novel variants. Overall, there was a high genetic heterogeneity, and the most frequent genetic diagnosis was SPG7 (n = 15), followed by SETX (n = 6), CACNA1A (n = 5), POLR3A (n = 4), and SYNE1 (n = 3). In addition, 17 families displayed likely pathogenic/pathogenic variants in 14 different genes: KCND3 (n = 2), KIF1C (n = 2), CYP27A1A (n = 2), AFG3L2 (n = 1), ANO10 (n = 1), CAPN1 (n = 1), CWF19L1 (n = 1), ITPR1 (n = 1), KCNA1 (n = 1), OPA1 (n = 1), PNPLA6 (n = 1), SPG11 (n = 1), SPTBN2 (n = 1), and TPP1 (n = 1). Twenty-two novel variants were characterized. SCA36 was diagnosed in 11 families, all with autosomal dominant (AD) presentation. SCA36 screening increased the total diagnostic rate to 47% (n = 61/130). Ultimately, undiagnosed patients showed delayed age at onset (p < 0.05) and were more frequently sporadic.
Discussion: Our study provides insight into the genetic landscape of CA in Eastern Spain. Although CES was an effective approach to capture genetic heterogeneity, most patients remained undiagnosed. SCA36 was found to be a relatively frequent form and, therefore, should be tested prior to CES in familial AD presentations in particular geographical regions.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
