Siti Aishah Abdul Wahab, Yusnita Yakob, Mohd Khairul Nizam Mohd Khalid, Noraishah Ali, Huey Yin Leong, Lock Hock Ngu
{"title":"Molecular, Biochemical, and Clinical Characterization of Thirteen Patients with Glycogen Storage Disease 1a in Malaysia.","authors":"Siti Aishah Abdul Wahab, Yusnita Yakob, Mohd Khairul Nizam Mohd Khalid, Noraishah Ali, Huey Yin Leong, Lock Hock Ngu","doi":"10.1155/2022/5870092","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Glycogen storage disease type 1a (GSD1a) is a rare autosomal recessive metabolic disorder characterized by hypoglycaemia, growth retardation, lactic acidosis, hepatomegaly, hyperlipidemia, and nephromegaly. GSD1a is caused by a mutation in the <i>G6PC</i> gene encoding glucose-6-phosphatase (G6Pase); an enzyme that catalyses the hydrolysis of glucose-6-phosphate (G6P) to phosphate and glucose.</p><p><strong>Objective: </strong>To elaborate on the clinical findings, biochemical data, molecular genetic analysis, and short-term prognosis of 13 GSD1a patients in Malaysia.</p><p><strong>Methods: </strong>The information about 13 clinically classified GSD1a patients was retrospectively studied. The <i>G6PC</i> mutation analysis was performed by PCR-DNA sequencing.</p><p><strong>Results: </strong>Patients were presented with hepatomegaly (92%), hypoglycaemia (38%), poor weight gain (23%), and short stature (15%). Mutation analysis revealed nine heterozygous mutations; eight previously reported mutations (c.155 A > T, c.209 G > A, c.226 A > T, c.248 G > A, c.648 G > T, c.706 T > A, c.1022 T > A, c.262delG) and a novel mutation (c.325 T > C). The most common mutation found in Malaysian patients was c.648 G > T in ten patients (77%) of mostly Malay ethnicity, followed by c.248 G > A in 4 patients of Chinese ethnicity (30%). A novel missense mutation (c.325 T > C) was predicted to be disease-causing by various <i>in silico</i> software.</p><p><strong>Conclusions: </strong>The establishment of <i>G6PC</i> molecular genetic testing will enable the detection of presymptomatic patients, assisting in genetic counselling while avoiding the invasive methods of liver biopsy.</p>","PeriodicalId":12778,"journal":{"name":"Genetics research","volume":"2022 ","pages":"5870092"},"PeriodicalIF":2.1000,"publicationDate":"2022-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9489408/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1155/2022/5870092","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Glycogen storage disease type 1a (GSD1a) is a rare autosomal recessive metabolic disorder characterized by hypoglycaemia, growth retardation, lactic acidosis, hepatomegaly, hyperlipidemia, and nephromegaly. GSD1a is caused by a mutation in the G6PC gene encoding glucose-6-phosphatase (G6Pase); an enzyme that catalyses the hydrolysis of glucose-6-phosphate (G6P) to phosphate and glucose.
Objective: To elaborate on the clinical findings, biochemical data, molecular genetic analysis, and short-term prognosis of 13 GSD1a patients in Malaysia.
Methods: The information about 13 clinically classified GSD1a patients was retrospectively studied. The G6PC mutation analysis was performed by PCR-DNA sequencing.
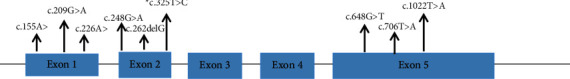
Results: Patients were presented with hepatomegaly (92%), hypoglycaemia (38%), poor weight gain (23%), and short stature (15%). Mutation analysis revealed nine heterozygous mutations; eight previously reported mutations (c.155 A > T, c.209 G > A, c.226 A > T, c.248 G > A, c.648 G > T, c.706 T > A, c.1022 T > A, c.262delG) and a novel mutation (c.325 T > C). The most common mutation found in Malaysian patients was c.648 G > T in ten patients (77%) of mostly Malay ethnicity, followed by c.248 G > A in 4 patients of Chinese ethnicity (30%). A novel missense mutation (c.325 T > C) was predicted to be disease-causing by various in silico software.
Conclusions: The establishment of G6PC molecular genetic testing will enable the detection of presymptomatic patients, assisting in genetic counselling while avoiding the invasive methods of liver biopsy.
期刊介绍:
Genetics Research is a key forum for original research on all aspects of human and animal genetics, reporting key findings on genomes, genes, mutations and molecular interactions, extending out to developmental, evolutionary, and population genetics as well as ethical, legal and social aspects. Our aim is to lead to a better understanding of genetic processes in health and disease. The journal focuses on the use of new technologies, such as next generation sequencing together with bioinformatics analysis, to produce increasingly detailed views of how genes function in tissues and how these genes perform, individually or collectively, in normal development and disease aetiology. The journal publishes original work, review articles, short papers, computational studies, and novel methods and techniques in research covering humans and well-established genetic organisms. Key subject areas include medical genetics, genomics, human evolutionary and population genetics, bioinformatics, genetics of complex traits, molecular and developmental genetics, Evo-Devo, quantitative and statistical genetics, behavioural genetics and environmental genetics. The breadth and quality of research make the journal an invaluable resource for medical geneticists, molecular biologists, bioinformaticians and researchers involved in genetic basis of diseases, evolutionary and developmental studies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
