J Shane Kippenhan, Michael D Gregory, Tiffany Nash, Philip Kohn, Carolyn B Mervis, Daniel P Eisenberg, Madeline H Garvey, Katherine Roe, Colleen A Morris, Bhaskar Kolachana, Ariel M Pani, Leah Sorcher, Karen F Berman
{"title":"Dorsal visual stream and LIMK1: hemideletion, haplotype, and enduring effects in children with Williams syndrome.","authors":"J Shane Kippenhan, Michael D Gregory, Tiffany Nash, Philip Kohn, Carolyn B Mervis, Daniel P Eisenberg, Madeline H Garvey, Katherine Roe, Colleen A Morris, Bhaskar Kolachana, Ariel M Pani, Leah Sorcher, Karen F Berman","doi":"10.1186/s11689-023-09493-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Williams syndrome (WS), a rare neurodevelopmental disorder caused by hemizygous deletion of ~ 25 genes from chromosomal band 7q11.23, affords an exceptional opportunity to study associations between a well-delineated genetic abnormality and a well-characterized neurobehavioral profile. Clinically, WS is typified by increased social drive (often termed \"hypersociability\") and severe visuospatial construction deficits. Previous studies have linked visuospatial problems in WS with alterations in the dorsal visual processing stream. We investigated the impacts of hemideletion and haplotype variation of LIMK1, a gene hemideleted in WS and linked to neuronal maturation and migration, on the structure and function of the dorsal stream, specifically the intraparietal sulcus (IPS), a region known to be altered in adults with WS.</p><p><strong>Methods: </strong>We tested for IPS structural and functional changes using longitudinal MRI in a developing cohort of children with WS (76 visits from 33 participants, compared to 280 visits from 94 typically developing age- and sex-matched participants) over the age range of 5-22. We also performed MRI studies of 12 individuals with rare, shorter hemideletions at 7q11.23, all of which included LIMK1. Finally, we tested for effects of LIMK1 variation on IPS structure and imputed LIMK1 expression in two independent cohorts of healthy individuals from the general population.</p><p><strong>Results: </strong>IPS structural (p < 10<sup>-4</sup> FDR corrected) and functional (p < .05 FDR corrected) anomalies previously reported in adults were confirmed in children with WS, and, consistent with an enduring genetic mechanism, were stable from early childhood into adulthood. In the short hemideletion cohort, IPS deficits similar to those in WS were found, although effect sizes were smaller than those found in WS for both structural and functional findings. Finally, in each of the two general population cohorts stratified by LIMK1 haplotype, IPS gray matter volume (p<sub>discovery</sub> < 0.05 SVC, p<sub>replication</sub> = 0.0015) and imputed LIMK1 expression (p<sub>discovery</sub> = 10<sup>-15</sup>, p<sub>replication</sub> = 10<sup>-23</sup>) varied according to LIMK1 haplotype.</p><p><strong>Conclusions: </strong>This work offers insight into neurobiological and genetic mechanisms responsible for the WS phenotype and also more generally provides a striking example of the mechanisms by which genetic variation, acting by means of molecular effects on a neural intermediary, can influence human cognition and, in some cases, lead to neurocognitive disorders.</p>","PeriodicalId":16530,"journal":{"name":"Journal of Neurodevelopmental Disorders","volume":"15 1","pages":"29"},"PeriodicalIF":4.0000,"publicationDate":"2023-08-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10464045/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Neurodevelopmental Disorders","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s11689-023-09493-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Williams syndrome (WS), a rare neurodevelopmental disorder caused by hemizygous deletion of ~ 25 genes from chromosomal band 7q11.23, affords an exceptional opportunity to study associations between a well-delineated genetic abnormality and a well-characterized neurobehavioral profile. Clinically, WS is typified by increased social drive (often termed "hypersociability") and severe visuospatial construction deficits. Previous studies have linked visuospatial problems in WS with alterations in the dorsal visual processing stream. We investigated the impacts of hemideletion and haplotype variation of LIMK1, a gene hemideleted in WS and linked to neuronal maturation and migration, on the structure and function of the dorsal stream, specifically the intraparietal sulcus (IPS), a region known to be altered in adults with WS.
Methods: We tested for IPS structural and functional changes using longitudinal MRI in a developing cohort of children with WS (76 visits from 33 participants, compared to 280 visits from 94 typically developing age- and sex-matched participants) over the age range of 5-22. We also performed MRI studies of 12 individuals with rare, shorter hemideletions at 7q11.23, all of which included LIMK1. Finally, we tested for effects of LIMK1 variation on IPS structure and imputed LIMK1 expression in two independent cohorts of healthy individuals from the general population.
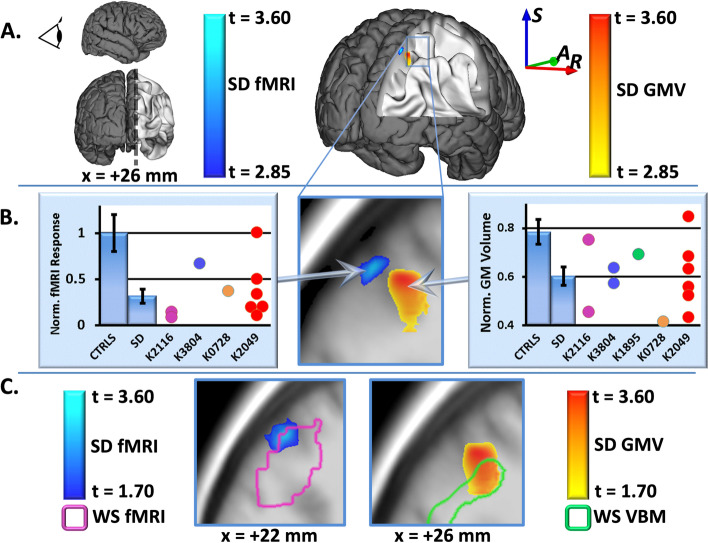
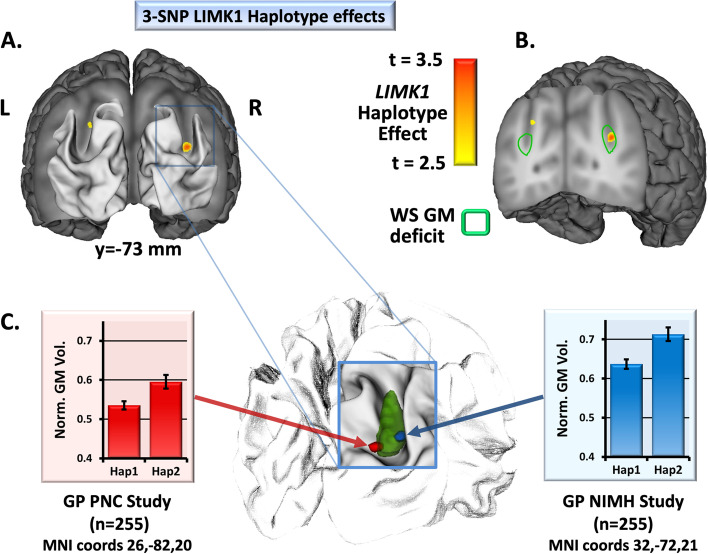
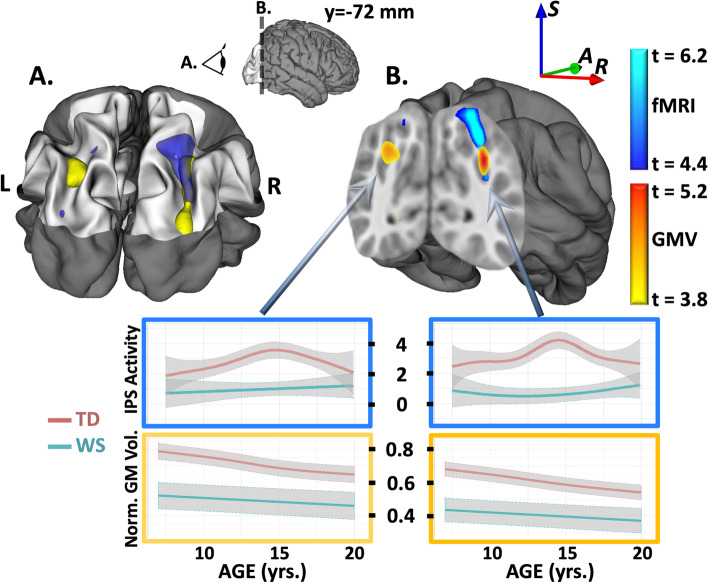
Results: IPS structural (p < 10-4 FDR corrected) and functional (p < .05 FDR corrected) anomalies previously reported in adults were confirmed in children with WS, and, consistent with an enduring genetic mechanism, were stable from early childhood into adulthood. In the short hemideletion cohort, IPS deficits similar to those in WS were found, although effect sizes were smaller than those found in WS for both structural and functional findings. Finally, in each of the two general population cohorts stratified by LIMK1 haplotype, IPS gray matter volume (pdiscovery < 0.05 SVC, preplication = 0.0015) and imputed LIMK1 expression (pdiscovery = 10-15, preplication = 10-23) varied according to LIMK1 haplotype.
Conclusions: This work offers insight into neurobiological and genetic mechanisms responsible for the WS phenotype and also more generally provides a striking example of the mechanisms by which genetic variation, acting by means of molecular effects on a neural intermediary, can influence human cognition and, in some cases, lead to neurocognitive disorders.
期刊介绍:
Journal of Neurodevelopmental Disorders is an open access journal that integrates current, cutting-edge research across a number of disciplines, including neurobiology, genetics, cognitive neuroscience, psychiatry and psychology. The journal’s primary focus is on the pathogenesis of neurodevelopmental disorders including autism, fragile X syndrome, tuberous sclerosis, Turner Syndrome, 22q Deletion Syndrome, Prader-Willi and Angelman Syndrome, Williams syndrome, lysosomal storage diseases, dyslexia, specific language impairment and fetal alcohol syndrome. With the discovery of specific genes underlying neurodevelopmental syndromes, the emergence of powerful tools for studying neural circuitry, and the development of new approaches for exploring molecular mechanisms, interdisciplinary research on the pathogenesis of neurodevelopmental disorders is now increasingly common. Journal of Neurodevelopmental Disorders provides a unique venue for researchers interested in comparing and contrasting mechanisms and characteristics related to the pathogenesis of the full range of neurodevelopmental disorders, sharpening our understanding of the etiology and relevant phenotypes of each condition.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
