Inhibition of mitochondrial fatty acid β-oxidation activates mTORC1 pathway and protein synthesis via Gcn5-dependent acetylation of Raptor in zebrafish.
Wen-Hao Zhou, Yuan Luo, Rui-Xin Li, Pascal Degrace, Tony Jourdan, Fang Qiao, Li-Qiao Chen, Mei-Ling Zhang, Zhen-Yu Du
{"title":"Inhibition of mitochondrial fatty acid β-oxidation activates mTORC1 pathway and protein synthesis via Gcn5-dependent acetylation of Raptor in zebrafish.","authors":"Wen-Hao Zhou, Yuan Luo, Rui-Xin Li, Pascal Degrace, Tony Jourdan, Fang Qiao, Li-Qiao Chen, Mei-Ling Zhang, Zhen-Yu Du","doi":"10.1016/j.jbc.2023.105220","DOIUrl":null,"url":null,"abstract":"<p><p>Pharmacological inhibition of mitochondrial fatty acid oxidation (FAO) has been clinically used to alleviate certain metabolic diseases by remodeling cellular metabolism. However, mitochondrial FAO inhibition also leads to mechanistic target of rapamycin complex 1 (mTORC1) activation-related protein synthesis and tissue hypertrophy, but the mechanism remains unclear. Here, by using a mitochondrial FAO inhibitor (mildronate or etomoxir) or knocking out carnitine palmitoyltransferase-1, we revealed that mitochondrial FAO inhibition activated the mTORC1 pathway through general control nondepressible 5-dependent Raptor acetylation. Mitochondrial FAO inhibition significantly promoted glucose catabolism and increased intracellular acetyl-CoA levels. In response to the increased intracellular acetyl-CoA, acetyltransferase general control nondepressible 5 activated mTORC1 by catalyzing Raptor acetylation through direct interaction. Further investigation also screened Raptor deacetylase histone deacetylase class II and identified histone deacetylase 7 as a potential regulator of Raptor. These results provide a possible mechanistic explanation for the mTORC1 activation after mitochondrial FAO inhibition and also bring light to reveal the roles of nutrient metabolic remodeling in regulating protein acetylation by affecting acetyl-CoA production.</p>","PeriodicalId":22621,"journal":{"name":"The Journal of Biological Chemistry","volume":" ","pages":"105220"},"PeriodicalIF":0.0000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10540046/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Biological Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.jbc.2023.105220","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/3 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
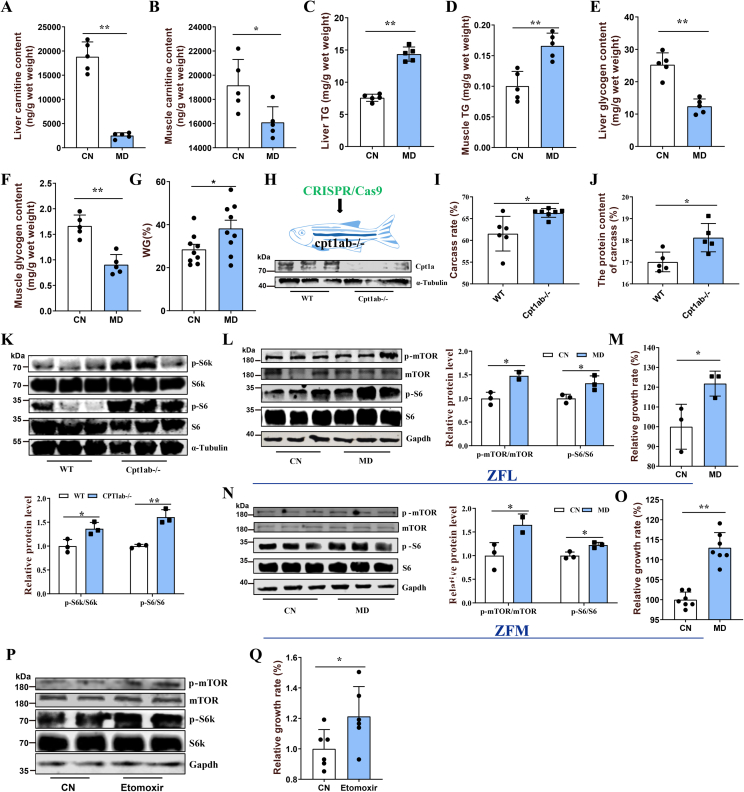
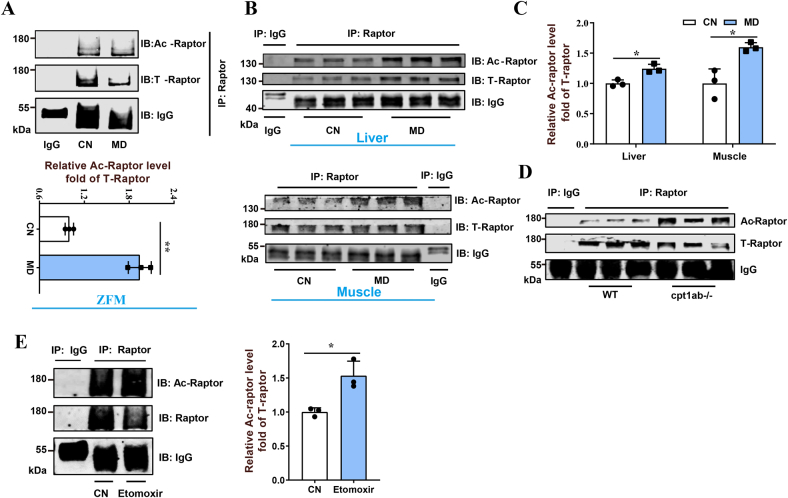
Pharmacological inhibition of mitochondrial fatty acid oxidation (FAO) has been clinically used to alleviate certain metabolic diseases by remodeling cellular metabolism. However, mitochondrial FAO inhibition also leads to mechanistic target of rapamycin complex 1 (mTORC1) activation-related protein synthesis and tissue hypertrophy, but the mechanism remains unclear. Here, by using a mitochondrial FAO inhibitor (mildronate or etomoxir) or knocking out carnitine palmitoyltransferase-1, we revealed that mitochondrial FAO inhibition activated the mTORC1 pathway through general control nondepressible 5-dependent Raptor acetylation. Mitochondrial FAO inhibition significantly promoted glucose catabolism and increased intracellular acetyl-CoA levels. In response to the increased intracellular acetyl-CoA, acetyltransferase general control nondepressible 5 activated mTORC1 by catalyzing Raptor acetylation through direct interaction. Further investigation also screened Raptor deacetylase histone deacetylase class II and identified histone deacetylase 7 as a potential regulator of Raptor. These results provide a possible mechanistic explanation for the mTORC1 activation after mitochondrial FAO inhibition and also bring light to reveal the roles of nutrient metabolic remodeling in regulating protein acetylation by affecting acetyl-CoA production.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
