Nodopathies and paranodopathies are autoimmune neuropathies associated with antibodies to nodal–paranodal antigens (neurofascin 140/186 and 155, contactin-1, contactin-associated protein 1 [Caspr1]) characterized by peculiar clinical features, poor response to standard immunotherapies (e.g., intravenous immunoglobulins, IVIg). Improvement after anti-CD20 monoclonal antibody therapy has been reported. Data on Caspr1 antibodies pathogenicity are still preliminary, and longitudinal titers have been poorly described.
We report on a young woman who developed a disabling neuropathy with antibodies to the Caspr1/contactin-1 complex showing a dramatic improvement after rituximab therapy, mirrored by the decrease of antibody titers.
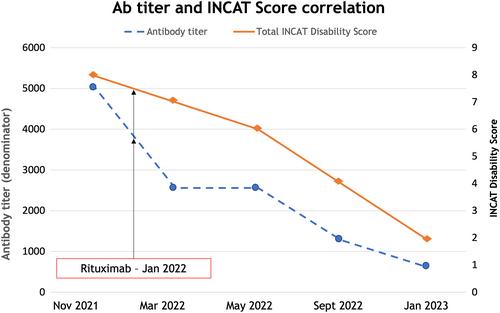
A 26-year-old woman presented with ataxic-stepping gait, severe motor weakness at four limbs, and low frequency postural tremor. For neurophysiological evidence of demyelinating neuropathy, she was diagnosed with chronic inflammatory demyelinating polyradiculoneuropathy and treated with IVIg without benefit. MRI showed symmetrical hypertrophy and marked signal hyperintensity of brachial and lumbosacral plexi. Cerebrospinal fluid showed 710 mg/dL protein. Despite intravenous methylprednisolone, the patient progressively worsened, and became wheelchair-bound. Antibodies to nodal–paranodal antigens were searched for by ELISA and cell-based assay. Anticontactin/Caspr1 IgG4 antibodies resulted positive. The patient underwent rituximab therapy with slow progressive improvement that mirrored the antibodies titer, measured throughout the disease course.
Our patient had a severe progressive course with early disability and axonal damage, and slow recovery starting only a few months after antibody-depleting therapy. The close correlation between titer, disability, and treatment, supports the pathogenicity of Caspr1 antibodies, and suggest that their longitudinal evaluation might provide a potential biomarker to evaluate treatment response.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
