Malindrie Dharmaratne, Ameya S Kulkarni, Atefeh Taherian Fard, Jessica C Mar
{"title":"scShapes: a statistical framework for identifying distribution shapes in single-cell RNA-sequencing data.","authors":"Malindrie Dharmaratne, Ameya S Kulkarni, Atefeh Taherian Fard, Jessica C Mar","doi":"10.1093/gigascience/giac126","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Single-cell RNA sequencing (scRNA-seq) methods have been advantageous for quantifying cell-to-cell variation by profiling the transcriptomes of individual cells. For scRNA-seq data, variability in gene expression reflects the degree of variation in gene expression from one cell to another. Analyses that focus on cell-cell variability therefore are useful for going beyond changes based on average expression and, instead, identifying genes with homogeneous expression versus those that vary widely from cell to cell.</p><p><strong>Results: </strong>We present a novel statistical framework, scShapes, for identifying differential distributions in single-cell RNA-sequencing data using generalized linear models. Most approaches for differential gene expression detect shifts in the mean value. However, as single-cell data are driven by overdispersion and dropouts, moving beyond means and using distributions that can handle excess zeros is critical. scShapes quantifies gene-specific cell-to-cell variability by testing for differences in the expression distribution while flexibly adjusting for covariates if required. We demonstrate that scShapes identifies subtle variations that are independent of altered mean expression and detects biologically relevant genes that were not discovered through standard approaches.</p><p><strong>Conclusions: </strong>This analysis also draws attention to genes that switch distribution shapes from a unimodal distribution to a zero-inflated distribution and raises open questions about the plausible biological mechanisms that may give rise to this, such as transcriptional bursting. Overall, the results from scShapes help to expand our understanding of the role that gene expression plays in the transcriptional regulation of a specific perturbation or cellular phenotype. Our framework scShapes is incorporated into a Bioconductor R package (https://www.bioconductor.org/packages/release/bioc/html/scShapes.html).</p>","PeriodicalId":12581,"journal":{"name":"GigaScience","volume":"12 ","pages":""},"PeriodicalIF":11.8000,"publicationDate":"2022-12-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9871437/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"GigaScience","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/gigascience/giac126","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/24 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
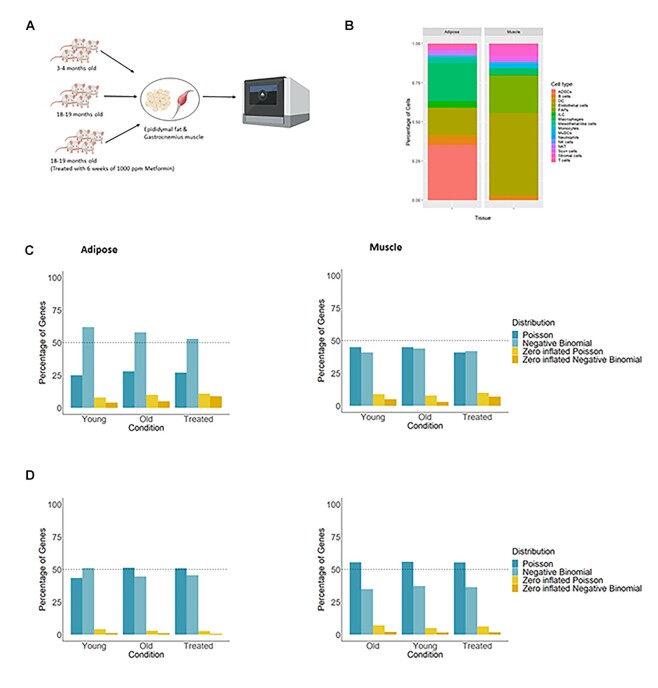
Background: Single-cell RNA sequencing (scRNA-seq) methods have been advantageous for quantifying cell-to-cell variation by profiling the transcriptomes of individual cells. For scRNA-seq data, variability in gene expression reflects the degree of variation in gene expression from one cell to another. Analyses that focus on cell-cell variability therefore are useful for going beyond changes based on average expression and, instead, identifying genes with homogeneous expression versus those that vary widely from cell to cell.
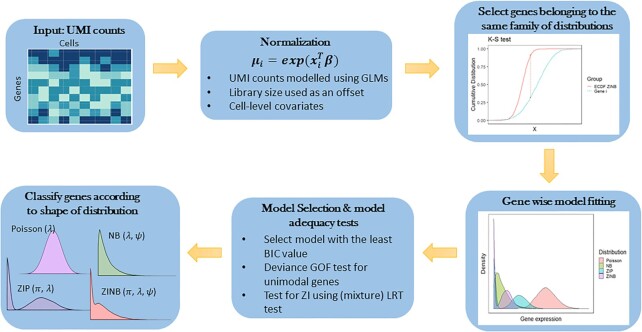
Results: We present a novel statistical framework, scShapes, for identifying differential distributions in single-cell RNA-sequencing data using generalized linear models. Most approaches for differential gene expression detect shifts in the mean value. However, as single-cell data are driven by overdispersion and dropouts, moving beyond means and using distributions that can handle excess zeros is critical. scShapes quantifies gene-specific cell-to-cell variability by testing for differences in the expression distribution while flexibly adjusting for covariates if required. We demonstrate that scShapes identifies subtle variations that are independent of altered mean expression and detects biologically relevant genes that were not discovered through standard approaches.
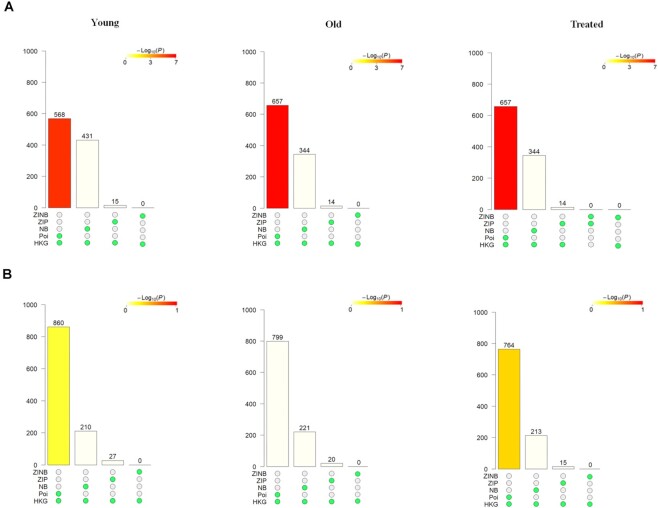
Conclusions: This analysis also draws attention to genes that switch distribution shapes from a unimodal distribution to a zero-inflated distribution and raises open questions about the plausible biological mechanisms that may give rise to this, such as transcriptional bursting. Overall, the results from scShapes help to expand our understanding of the role that gene expression plays in the transcriptional regulation of a specific perturbation or cellular phenotype. Our framework scShapes is incorporated into a Bioconductor R package (https://www.bioconductor.org/packages/release/bioc/html/scShapes.html).
期刊介绍:
GigaScience seeks to transform data dissemination and utilization in the life and biomedical sciences. As an online open-access open-data journal, it specializes in publishing "big-data" studies encompassing various fields. Its scope includes not only "omic" type data and the fields of high-throughput biology currently serviced by large public repositories, but also the growing range of more difficult-to-access data, such as imaging, neuroscience, ecology, cohort data, systems biology and other new types of large-scale shareable data.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
