Specific pathway abundances in the neonatal calf faecal microbiome are associated with susceptibility to Cryptosporidium parvum infection: a metagenomic analysis.
M F Hares, B E Griffiths, F Johnson, C Nelson, S Haldenby, C J Stewart, J S Duncan, G Oikonomou, J L Coombes
{"title":"Specific pathway abundances in the neonatal calf faecal microbiome are associated with susceptibility to Cryptosporidium parvum infection: a metagenomic analysis.","authors":"M F Hares, B E Griffiths, F Johnson, C Nelson, S Haldenby, C J Stewart, J S Duncan, G Oikonomou, J L Coombes","doi":"10.1186/s42523-023-00265-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cryptosporidium parvum is the main cause of calf scour worldwide. With limited therapeutic options and research compared to other Apicomplexa, it is important to understand the parasites' biology and interactions with the host and microbiome in order to develop novel strategies against this infection. The age-dependent nature of symptomatic cryptosporidiosis suggests a link to the undeveloped immune response, the immature intestinal epithelium, and its associated microbiota. This led us to hypothesise that specific features of the early life microbiome could predict calf susceptibility to C. parvum infection.</p><p><strong>Results: </strong>In this study, a single faecal swab sample was collected from each calf within the first week of life in a cohort of 346 animals. All 346 calves were subsequently monitored for clinical signs of cryptosporidiosis, and calves that developed diarrhoea were tested for Rotavirus, Coronavirus, E. coli F5 (K99) and C. parvum by lateral flow test (LFT). A retrospective case-control approach was taken whereby a subset of healthy calves (Control group; n = 33) and calves that went on to develop clinical signs of infectious diarrhoea and test positive for C. parvum infection via LFT (Cryptosporidium-positive group; n = 32) were selected from this cohort, five of which were excluded due to low DNA quality. A metagenomic analysis was conducted on the faecal microbiomes of the control group (n = 30) and the Cryptosporidium-positive group (n = 30) prior to infection, to determine features predictive of cryptosporidiosis. Taxonomic analysis showed no significant differences in alpha diversity, beta diversity, and taxa relative abundance between controls and Cryptosporidium-positive groups. Analysis of functional potential showed pathways related to isoprenoid precursor, haem and purine biosynthesis were significantly higher in abundance in calves that later tested positive for C. parvum (q ≤ 0.25). These pathways are either absent or streamlined in the C. parvum parasites. Though the de novo production of isoprenoid precursors, haem and purines are absent, C. parvum has been shown to encode enzymes that catalyse the downstream reactions of these pathway metabolites, indicating that C. parvum may scavenge those products from an external source.</p><p><strong>Conclusions: </strong>The host has previously been put forward as the source of essential metabolites, but our study suggests that C. parvum may also be able to harness specific metabolic pathways of the microbiota in order to survive and replicate. This finding is important as components of these microbial pathways could be exploited as potential therapeutic targets for the prevention or mitigation of cryptosporidiosis in bovine neonates.</p>","PeriodicalId":72201,"journal":{"name":"Animal microbiome","volume":"5 1","pages":"43"},"PeriodicalIF":4.4000,"publicationDate":"2023-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10496319/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Animal microbiome","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42523-023-00265-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Cryptosporidium parvum is the main cause of calf scour worldwide. With limited therapeutic options and research compared to other Apicomplexa, it is important to understand the parasites' biology and interactions with the host and microbiome in order to develop novel strategies against this infection. The age-dependent nature of symptomatic cryptosporidiosis suggests a link to the undeveloped immune response, the immature intestinal epithelium, and its associated microbiota. This led us to hypothesise that specific features of the early life microbiome could predict calf susceptibility to C. parvum infection.
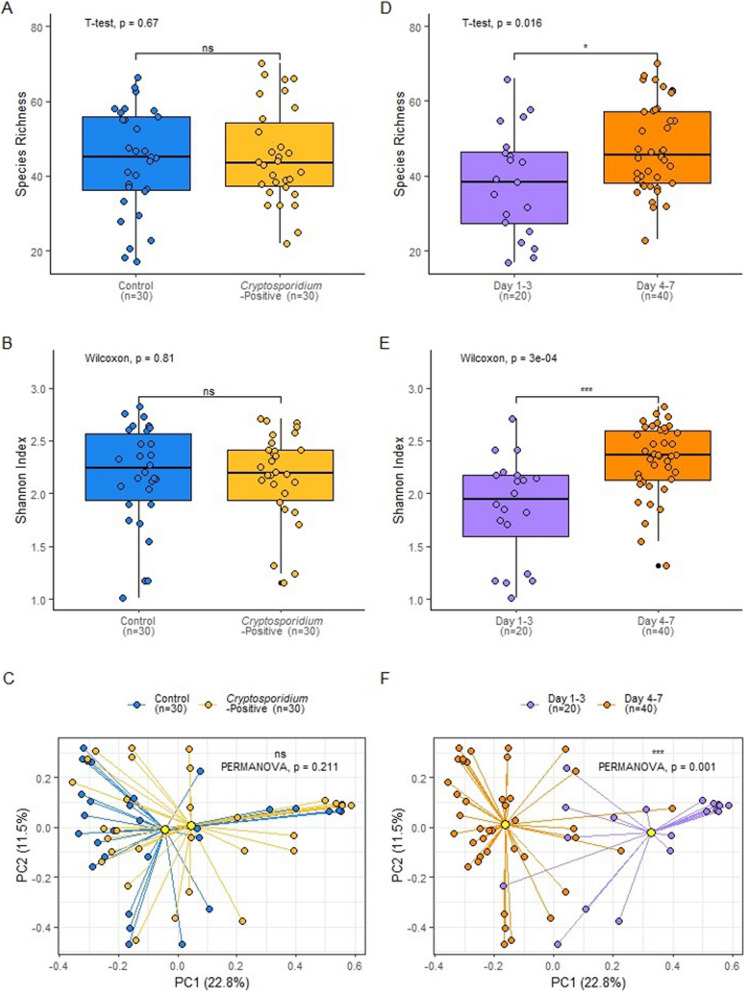
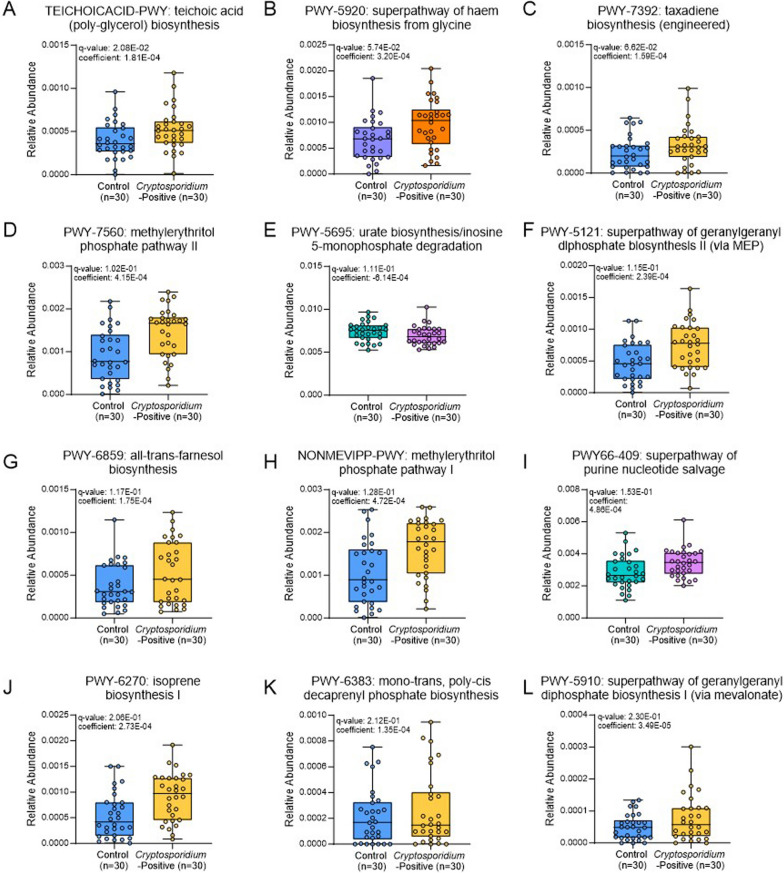
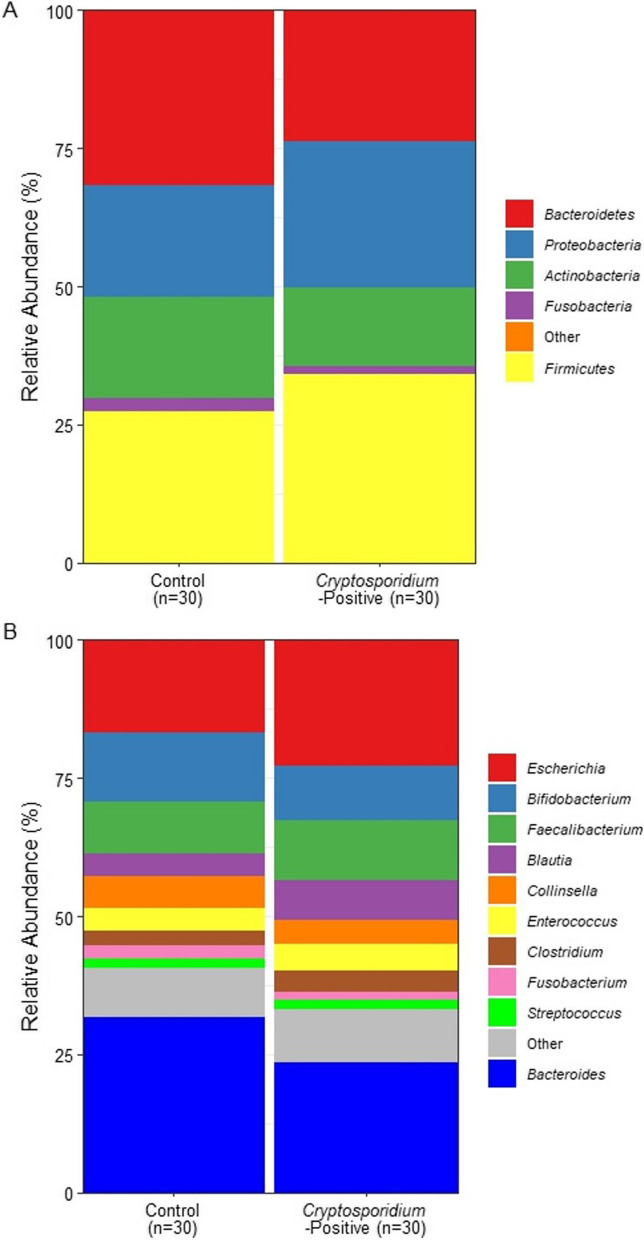
Results: In this study, a single faecal swab sample was collected from each calf within the first week of life in a cohort of 346 animals. All 346 calves were subsequently monitored for clinical signs of cryptosporidiosis, and calves that developed diarrhoea were tested for Rotavirus, Coronavirus, E. coli F5 (K99) and C. parvum by lateral flow test (LFT). A retrospective case-control approach was taken whereby a subset of healthy calves (Control group; n = 33) and calves that went on to develop clinical signs of infectious diarrhoea and test positive for C. parvum infection via LFT (Cryptosporidium-positive group; n = 32) were selected from this cohort, five of which were excluded due to low DNA quality. A metagenomic analysis was conducted on the faecal microbiomes of the control group (n = 30) and the Cryptosporidium-positive group (n = 30) prior to infection, to determine features predictive of cryptosporidiosis. Taxonomic analysis showed no significant differences in alpha diversity, beta diversity, and taxa relative abundance between controls and Cryptosporidium-positive groups. Analysis of functional potential showed pathways related to isoprenoid precursor, haem and purine biosynthesis were significantly higher in abundance in calves that later tested positive for C. parvum (q ≤ 0.25). These pathways are either absent or streamlined in the C. parvum parasites. Though the de novo production of isoprenoid precursors, haem and purines are absent, C. parvum has been shown to encode enzymes that catalyse the downstream reactions of these pathway metabolites, indicating that C. parvum may scavenge those products from an external source.
Conclusions: The host has previously been put forward as the source of essential metabolites, but our study suggests that C. parvum may also be able to harness specific metabolic pathways of the microbiota in order to survive and replicate. This finding is important as components of these microbial pathways could be exploited as potential therapeutic targets for the prevention or mitigation of cryptosporidiosis in bovine neonates.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
