{"title":"Phylogenetic inference using generative adversarial networks.","authors":"Megan L Smith, Matthew W Hahn","doi":"10.1093/bioinformatics/btad543","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>The application of machine learning approaches in phylogenetics has been impeded by the vast model space associated with inference. Supervised machine learning approaches require data from across this space to train models. Because of this, previous approaches have typically been limited to inferring relationships among unrooted quartets of taxa, where there are only three possible topologies. Here, we explore the potential of generative adversarial networks (GANs) to address this limitation. GANs consist of a generator and a discriminator: at each step, the generator aims to create data that is similar to real data, while the discriminator attempts to distinguish generated and real data. By using an evolutionary model as the generator, we use GANs to make evolutionary inferences. Since a new model can be considered at each iteration, heuristic searches of complex model spaces are possible. Thus, GANs offer a potential solution to the challenges of applying machine learning in phylogenetics.</p><p><strong>Results: </strong>We developed phyloGAN, a GAN that infers phylogenetic relationships among species. phyloGAN takes as input a concatenated alignment, or a set of gene alignments, and infers a phylogenetic tree either considering or ignoring gene tree heterogeneity. We explored the performance of phyloGAN for up to 15 taxa in the concatenation case and 6 taxa when considering gene tree heterogeneity. Error rates are relatively low in these simple cases. However, run times are slow and performance metrics suggest issues during training. Future work should explore novel architectures that may result in more stable and efficient GANs for phylogenetics.</p><p><strong>Availability and implementation: </strong>phyloGAN is available on github: https://github.com/meganlsmith/phyloGAN/.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10500083/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad543","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: The application of machine learning approaches in phylogenetics has been impeded by the vast model space associated with inference. Supervised machine learning approaches require data from across this space to train models. Because of this, previous approaches have typically been limited to inferring relationships among unrooted quartets of taxa, where there are only three possible topologies. Here, we explore the potential of generative adversarial networks (GANs) to address this limitation. GANs consist of a generator and a discriminator: at each step, the generator aims to create data that is similar to real data, while the discriminator attempts to distinguish generated and real data. By using an evolutionary model as the generator, we use GANs to make evolutionary inferences. Since a new model can be considered at each iteration, heuristic searches of complex model spaces are possible. Thus, GANs offer a potential solution to the challenges of applying machine learning in phylogenetics.
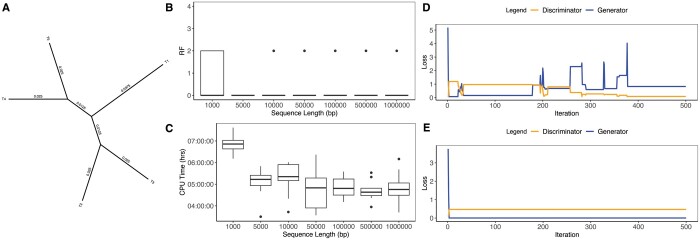
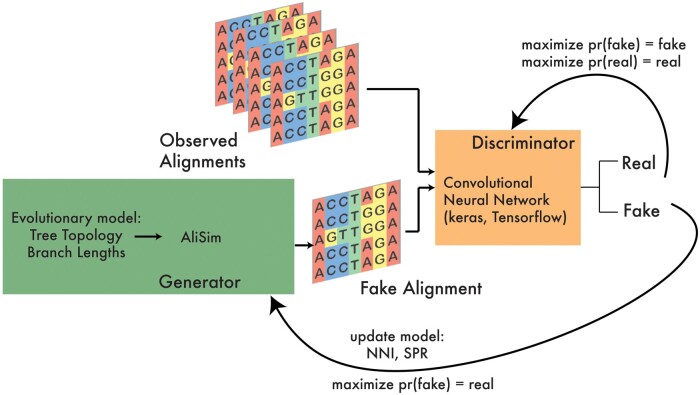
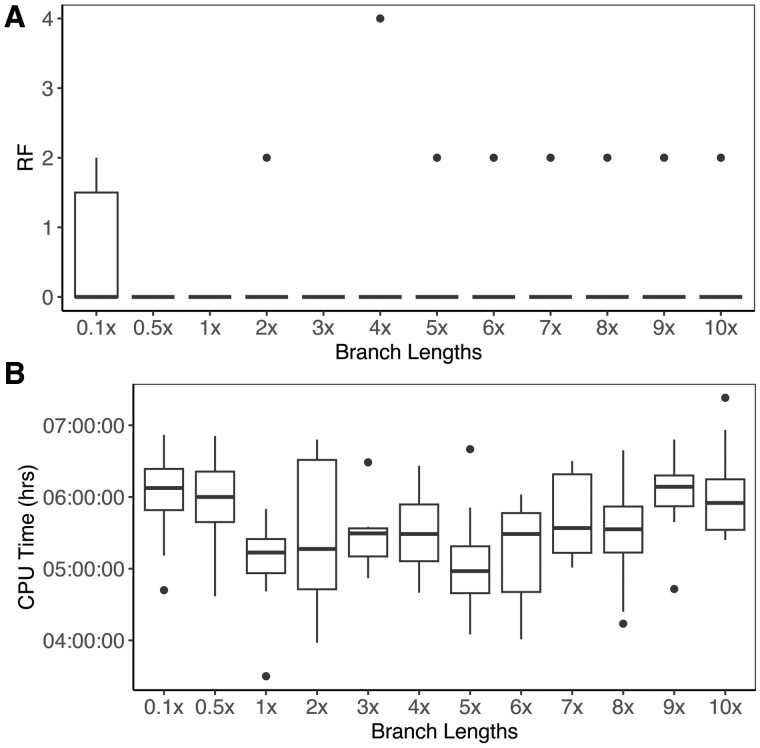
Results: We developed phyloGAN, a GAN that infers phylogenetic relationships among species. phyloGAN takes as input a concatenated alignment, or a set of gene alignments, and infers a phylogenetic tree either considering or ignoring gene tree heterogeneity. We explored the performance of phyloGAN for up to 15 taxa in the concatenation case and 6 taxa when considering gene tree heterogeneity. Error rates are relatively low in these simple cases. However, run times are slow and performance metrics suggest issues during training. Future work should explore novel architectures that may result in more stable and efficient GANs for phylogenetics.
Availability and implementation: phyloGAN is available on github: https://github.com/meganlsmith/phyloGAN/.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
