{"title":"Multimodal learning of noncoding variant effects using genome sequence and chromatin structure.","authors":"Wuwei Tan, Yang Shen","doi":"10.1093/bioinformatics/btad541","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>A growing amount of noncoding genetic variants, including single-nucleotide polymorphisms, are found to be associated with complex human traits and diseases. Their mechanistic interpretation is relatively limited and can use the help from computational prediction of their effects on epigenetic profiles. However, current models often focus on local, 1D genome sequence determinants and disregard global, 3D chromatin structure that critically affects epigenetic events.</p><p><strong>Results: </strong>We find that noncoding variants of unexpected high similarity in epigenetic profiles, with regards to their relatively low similarity in local sequences, can be largely attributed to their proximity in chromatin structure. Accordingly, we have developed a multimodal deep learning scheme that incorporates both data of 1D genome sequence and 3D chromatin structure for predicting noncoding variant effects. Specifically, we have integrated convolutional and recurrent neural networks for sequence embedding and graph neural networks for structure embedding despite the resolution gap between the two types of data, while utilizing recent DNA language models. Numerical results show that our models outperform competing sequence-only models in predicting epigenetic profiles and their use of long-range interactions complement sequence-only models in extracting regulatory motifs. They prove to be excellent predictors for noncoding variant effects in gene expression and pathogenicity, whether in unsupervised \"zero-shot\" learning or supervised \"few-shot\" learning.</p><p><strong>Availability and implementation: </strong>Codes and data can be accessed at https://github.com/Shen-Lab/ncVarPred-1D3D and https://zenodo.org/record/7975777.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10502240/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad541","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: A growing amount of noncoding genetic variants, including single-nucleotide polymorphisms, are found to be associated with complex human traits and diseases. Their mechanistic interpretation is relatively limited and can use the help from computational prediction of their effects on epigenetic profiles. However, current models often focus on local, 1D genome sequence determinants and disregard global, 3D chromatin structure that critically affects epigenetic events.
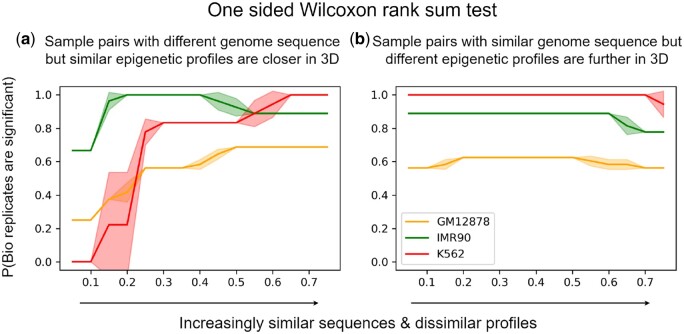
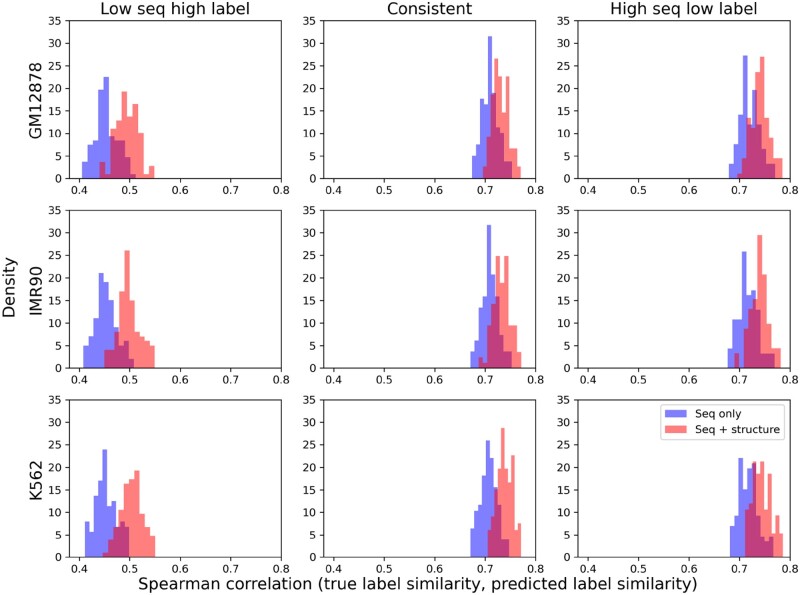
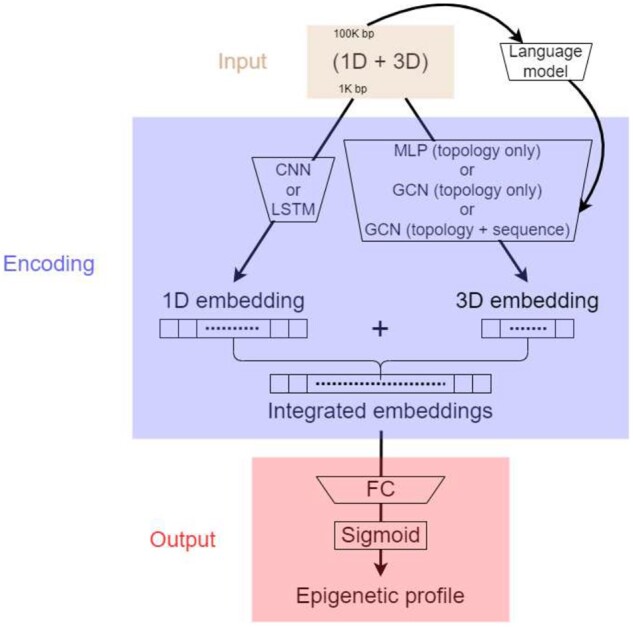
Results: We find that noncoding variants of unexpected high similarity in epigenetic profiles, with regards to their relatively low similarity in local sequences, can be largely attributed to their proximity in chromatin structure. Accordingly, we have developed a multimodal deep learning scheme that incorporates both data of 1D genome sequence and 3D chromatin structure for predicting noncoding variant effects. Specifically, we have integrated convolutional and recurrent neural networks for sequence embedding and graph neural networks for structure embedding despite the resolution gap between the two types of data, while utilizing recent DNA language models. Numerical results show that our models outperform competing sequence-only models in predicting epigenetic profiles and their use of long-range interactions complement sequence-only models in extracting regulatory motifs. They prove to be excellent predictors for noncoding variant effects in gene expression and pathogenicity, whether in unsupervised "zero-shot" learning or supervised "few-shot" learning.
Availability and implementation: Codes and data can be accessed at https://github.com/Shen-Lab/ncVarPred-1D3D and https://zenodo.org/record/7975777.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
