Megan E Forrest, Alayne P Meyer, Stephanie M Laureano Figueroa, Anthony Antonellis
{"title":"A missense, loss-of-function <i>YARS1</i> variant in a patient with proximal-predominant motor neuropathy.","authors":"Megan E Forrest, Alayne P Meyer, Stephanie M Laureano Figueroa, Anthony Antonellis","doi":"10.1101/mcs.a006246","DOIUrl":null,"url":null,"abstract":"<p><p>Aminoacyl-tRNA synthetases (ARSs) are essential enzymes with a critical role in protein synthesis: charging tRNA molecules with cognate amino acids. Heterozygosity for variants in five genes (<i>AARS1</i>, <i>GARS1</i>, <i>HARS1</i>, <i>WARS1</i>, and <i>YARS1</i>) encoding cytoplasmic, dimeric ARSs have been associated with autosomal dominant neurological phenotypes, including axonal Charcot-Marie-Tooth disease (CMT). Missense variants in the catalytic domain of <i>YARS1</i> were previously linked to dominant intermediate CMT type C (DI-CMTC). Here, we report a patient with a missense variant of unknown significance predicted to modify residue 308 in the anticodon binding domain of <i>YARS1</i> (p.Asp308Tyr). Interestingly, p.Asp308Tyr is associated with proximal-predominant motor neuropathy, which has not been reported in patients with pathogenic <i>YARS1</i> variants. We demonstrate that this allele causes a loss-of-function effect in yeast complementation assays when modeled in <i>YARS1</i> and the yeast ortholog <i>TYS1</i>; structural modeling of this variant further supports a loss-of-function effect. Taken together, this study raises the possibility that certain <i>YARS1</i> variants cause proximal-prominent motor neuropathy and indicates that patients with this phenotype should be screened for genetic lesions in <i>YARS1</i>.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":"8 7","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2022-12-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0e/04/MCS006246For.PMC9808560.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006246","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/12/1 0:00:00","PubModel":"Print","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
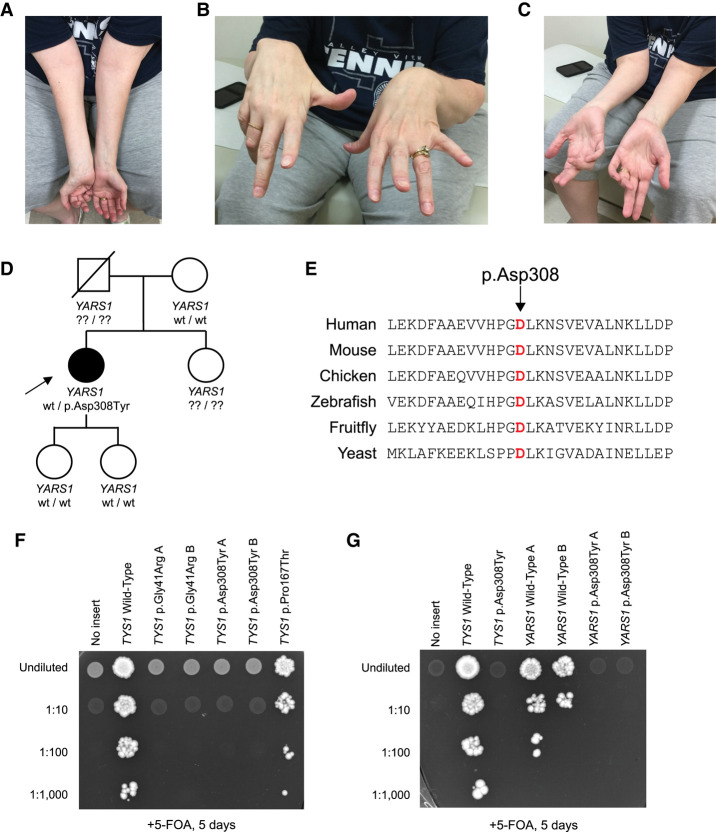
Aminoacyl-tRNA synthetases (ARSs) are essential enzymes with a critical role in protein synthesis: charging tRNA molecules with cognate amino acids. Heterozygosity for variants in five genes (AARS1, GARS1, HARS1, WARS1, and YARS1) encoding cytoplasmic, dimeric ARSs have been associated with autosomal dominant neurological phenotypes, including axonal Charcot-Marie-Tooth disease (CMT). Missense variants in the catalytic domain of YARS1 were previously linked to dominant intermediate CMT type C (DI-CMTC). Here, we report a patient with a missense variant of unknown significance predicted to modify residue 308 in the anticodon binding domain of YARS1 (p.Asp308Tyr). Interestingly, p.Asp308Tyr is associated with proximal-predominant motor neuropathy, which has not been reported in patients with pathogenic YARS1 variants. We demonstrate that this allele causes a loss-of-function effect in yeast complementation assays when modeled in YARS1 and the yeast ortholog TYS1; structural modeling of this variant further supports a loss-of-function effect. Taken together, this study raises the possibility that certain YARS1 variants cause proximal-prominent motor neuropathy and indicates that patients with this phenotype should be screened for genetic lesions in YARS1.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
