Xinwei He, Kun Qian, Ziqian Wang, Shirou Zeng, Hongwei Li, Wei Vivian Li
{"title":"scAce: an adaptive embedding and clustering method for single-cell gene expression data.","authors":"Xinwei He, Kun Qian, Ziqian Wang, Shirou Zeng, Hongwei Li, Wei Vivian Li","doi":"10.1093/bioinformatics/btad546","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Since the development of single-cell RNA sequencing (scRNA-seq) technologies, clustering analysis of single-cell gene expression data has been an essential tool for distinguishing cell types and identifying novel cell types. Even though many methods have been available for scRNA-seq clustering analysis, the majority of them are constrained by the requirement on predetermined cluster numbers or the dependence on selected initial cluster assignment.</p><p><strong>Results: </strong>In this article, we propose an adaptive embedding and clustering method named scAce, which constructs a variational autoencoder to simultaneously learn cell embeddings and cluster assignments. In the scAce method, we develop an adaptive cluster merging approach which achieves improved clustering results without the need to estimate the number of clusters in advance. In addition, scAce provides an option to perform clustering enhancement, which can update and enhance cluster assignments based on previous clustering results from other methods. Based on computational analysis of both simulated and real datasets, we demonstrate that scAce outperforms state-of-the-art clustering methods for scRNA-seq data, and achieves better clustering accuracy and robustness.</p><p><strong>Availability and implementation: </strong>The scAce package is implemented in python 3.8 and is freely available from https://github.com/sldyns/scAce.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10500084/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad546","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Since the development of single-cell RNA sequencing (scRNA-seq) technologies, clustering analysis of single-cell gene expression data has been an essential tool for distinguishing cell types and identifying novel cell types. Even though many methods have been available for scRNA-seq clustering analysis, the majority of them are constrained by the requirement on predetermined cluster numbers or the dependence on selected initial cluster assignment.
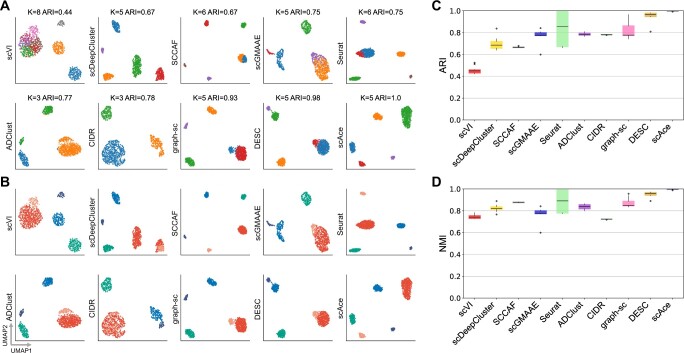
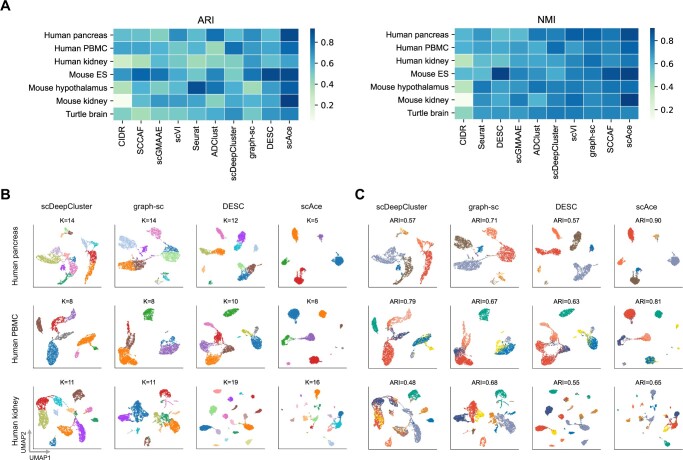
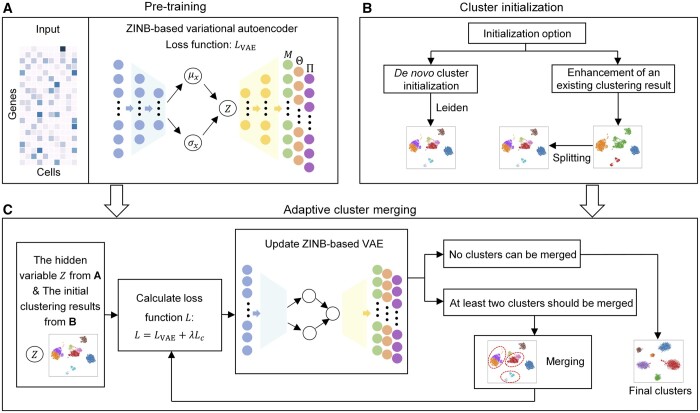
Results: In this article, we propose an adaptive embedding and clustering method named scAce, which constructs a variational autoencoder to simultaneously learn cell embeddings and cluster assignments. In the scAce method, we develop an adaptive cluster merging approach which achieves improved clustering results without the need to estimate the number of clusters in advance. In addition, scAce provides an option to perform clustering enhancement, which can update and enhance cluster assignments based on previous clustering results from other methods. Based on computational analysis of both simulated and real datasets, we demonstrate that scAce outperforms state-of-the-art clustering methods for scRNA-seq data, and achieves better clustering accuracy and robustness.
Availability and implementation: The scAce package is implemented in python 3.8 and is freely available from https://github.com/sldyns/scAce.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
