Georgi K Kanev, Yaran Zhang, Albert J Kooistra, Andreas Bender, Rob Leurs, David Bailey, Thomas Würdinger, Chris de Graaf, Iwan J P de Esch, Bart A Westerman
{"title":"Predicting the target landscape of kinase inhibitors using 3D convolutional neural networks.","authors":"Georgi K Kanev, Yaran Zhang, Albert J Kooistra, Andreas Bender, Rob Leurs, David Bailey, Thomas Würdinger, Chris de Graaf, Iwan J P de Esch, Bart A Westerman","doi":"10.1371/journal.pcbi.1011301","DOIUrl":null,"url":null,"abstract":"<p><p>Many therapies in clinical trials are based on single drug-single target relationships. To further extend this concept to multi-target approaches using multi-targeted drugs, we developed a machine learning pipeline to unravel the target landscape of kinase inhibitors. This pipeline, which we call 3D-KINEssence, uses a new type of protein fingerprints (3D FP) based on the structure of kinases generated through a 3D convolutional neural network (3D-CNN). These 3D-CNN kinase fingerprints were matched to molecular Morgan fingerprints to predict the targets of each respective kinase inhibitor based on available bioactivity data. The performance of the pipeline was evaluated on two test sets: a sparse drug-target set where each drug is matched in most cases to a single target and also on a densely-covered drug-target set where each drug is matched to most if not all targets. This latter set is more challenging to train, given its non-exclusive character. Our model's root-mean-square error (RMSE) based on the two datasets was 0.68 and 0.8, respectively. These results indicate that 3D FP can predict the target landscape of kinase inhibitors at around 0.8 log units of bioactivity. Our strategy can be utilized in proteochemometric or chemogenomic workflows by consolidating the target landscape of kinase inhibitors.</p>","PeriodicalId":49688,"journal":{"name":"PLoS Computational Biology","volume":"19 9","pages":"e1011301"},"PeriodicalIF":3.6000,"publicationDate":"2023-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10508635/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PLoS Computational Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1371/journal.pcbi.1011301","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
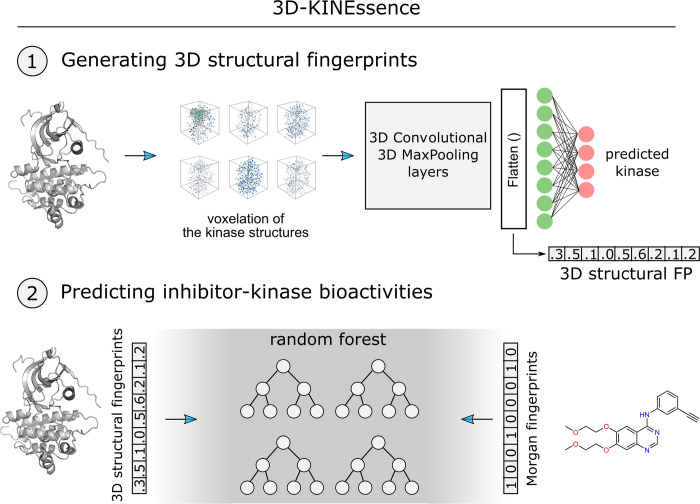
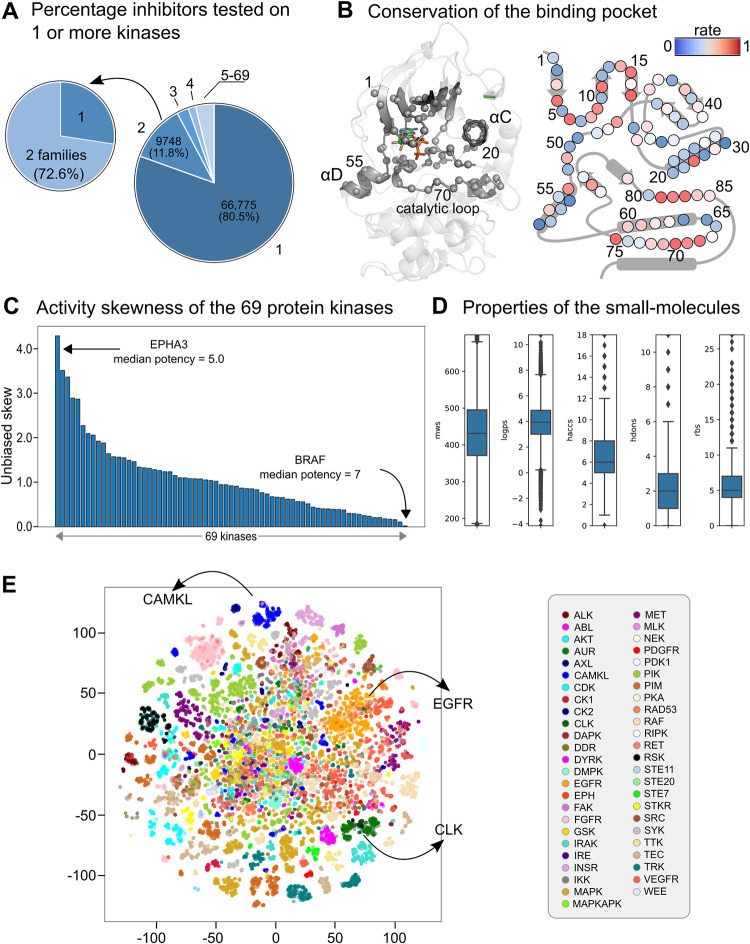
Many therapies in clinical trials are based on single drug-single target relationships. To further extend this concept to multi-target approaches using multi-targeted drugs, we developed a machine learning pipeline to unravel the target landscape of kinase inhibitors. This pipeline, which we call 3D-KINEssence, uses a new type of protein fingerprints (3D FP) based on the structure of kinases generated through a 3D convolutional neural network (3D-CNN). These 3D-CNN kinase fingerprints were matched to molecular Morgan fingerprints to predict the targets of each respective kinase inhibitor based on available bioactivity data. The performance of the pipeline was evaluated on two test sets: a sparse drug-target set where each drug is matched in most cases to a single target and also on a densely-covered drug-target set where each drug is matched to most if not all targets. This latter set is more challenging to train, given its non-exclusive character. Our model's root-mean-square error (RMSE) based on the two datasets was 0.68 and 0.8, respectively. These results indicate that 3D FP can predict the target landscape of kinase inhibitors at around 0.8 log units of bioactivity. Our strategy can be utilized in proteochemometric or chemogenomic workflows by consolidating the target landscape of kinase inhibitors.
期刊介绍:
PLOS Computational Biology features works of exceptional significance that further our understanding of living systems at all scales—from molecules and cells, to patient populations and ecosystems—through the application of computational methods. Readers include life and computational scientists, who can take the important findings presented here to the next level of discovery.
Research articles must be declared as belonging to a relevant section. More information about the sections can be found in the submission guidelines.
Research articles should model aspects of biological systems, demonstrate both methodological and scientific novelty, and provide profound new biological insights.
Generally, reliability and significance of biological discovery through computation should be validated and enriched by experimental studies. Inclusion of experimental validation is not required for publication, but should be referenced where possible. Inclusion of experimental validation of a modest biological discovery through computation does not render a manuscript suitable for PLOS Computational Biology.
Research articles specifically designated as Methods papers should describe outstanding methods of exceptional importance that have been shown, or have the promise to provide new biological insights. The method must already be widely adopted, or have the promise of wide adoption by a broad community of users. Enhancements to existing published methods will only be considered if those enhancements bring exceptional new capabilities.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
