Daniel James Clark, Thomas Murray, Michael Drees, Neil Kulkarni
{"title":"A Case of ALG6-CDG with Explosive Onset of Intractable Epilepsy During Infancy.","authors":"Daniel James Clark, Thomas Murray, Michael Drees, Neil Kulkarni","doi":"10.1177/2329048X231153781","DOIUrl":null,"url":null,"abstract":"<p><p>ALG6-CDG is a rare, but second most common, type 1 congenital disorder of glycosylation (CDG) caused by a defect in the α-1-3-glucosyltransferase (ALG6) enzyme in the N-glycan assembly pathway. Many mutations have been identified and inherited in an autosomal recessive pattern. There are less than 100 ALG6-CDG cases reported, all sharing the phenotype of hypotonia and developmental delay. The majority (perhaps >70%) have seizures, but a minority have intractable epilepsy or epileptic encephalopathy. We report the clinical course, EEG findings, and neuroimaging of a child found to have compound heterozygous alleles c.257 + 5G > A and c.680G > A (p.G227E) who developed explosive onset of intractable epilepsy and epileptic encephalopathy. Initially, CDG was not suspected due to its rarity and lack of multi-organ system involvement, but rapid whole exam sequence (8-day turnaround) revealed the specific diagnosis quickly.</p>","PeriodicalId":72572,"journal":{"name":"Child neurology open","volume":"10 ","pages":"2329048X231153781"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/cf/10.1177_2329048X231153781.PMC9900650.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Child neurology open","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/2329048X231153781","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
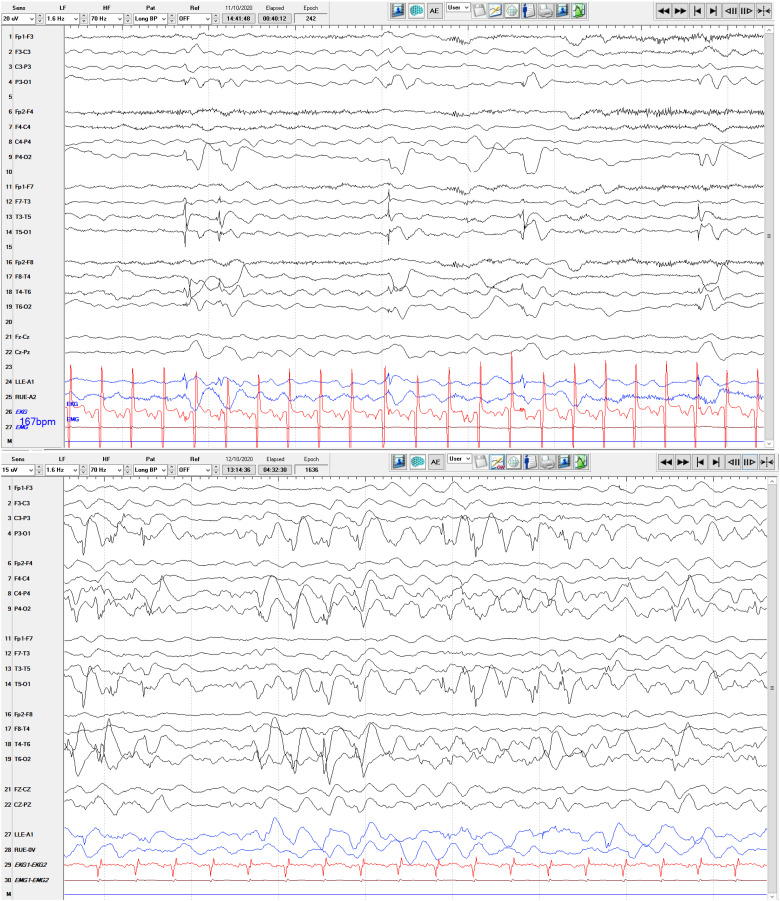
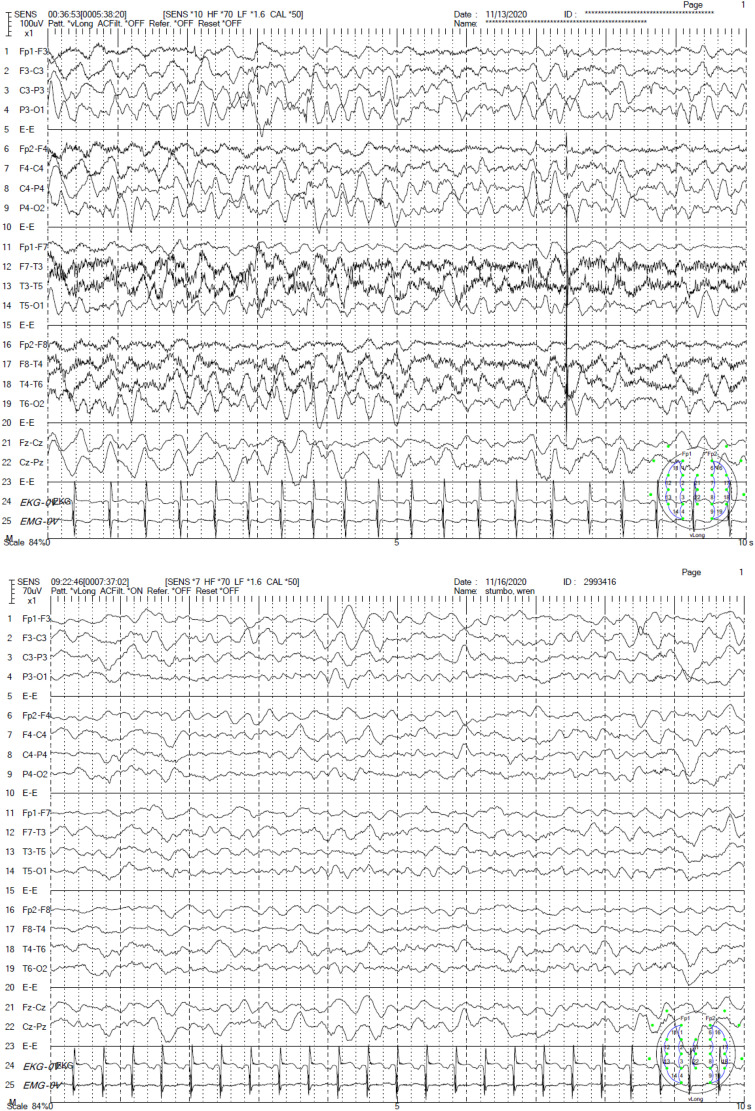
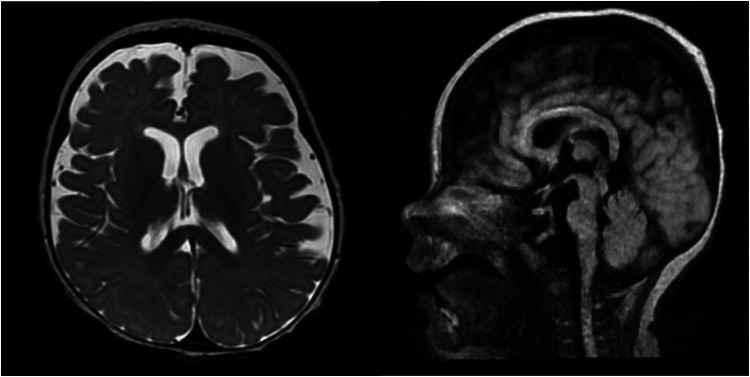
ALG6-CDG is a rare, but second most common, type 1 congenital disorder of glycosylation (CDG) caused by a defect in the α-1-3-glucosyltransferase (ALG6) enzyme in the N-glycan assembly pathway. Many mutations have been identified and inherited in an autosomal recessive pattern. There are less than 100 ALG6-CDG cases reported, all sharing the phenotype of hypotonia and developmental delay. The majority (perhaps >70%) have seizures, but a minority have intractable epilepsy or epileptic encephalopathy. We report the clinical course, EEG findings, and neuroimaging of a child found to have compound heterozygous alleles c.257 + 5G > A and c.680G > A (p.G227E) who developed explosive onset of intractable epilepsy and epileptic encephalopathy. Initially, CDG was not suspected due to its rarity and lack of multi-organ system involvement, but rapid whole exam sequence (8-day turnaround) revealed the specific diagnosis quickly.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
