David N Nicholson, Daniel S Himmelstein, Casey S Greene
{"title":"Expanding a database-derived biomedical knowledge graph via multi-relation extraction from biomedical abstracts.","authors":"David N Nicholson, Daniel S Himmelstein, Casey S Greene","doi":"10.1186/s13040-022-00311-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Knowledge graphs support biomedical research efforts by providing contextual information for biomedical entities, constructing networks, and supporting the interpretation of high-throughput analyses. These databases are populated via manual curation, which is challenging to scale with an exponentially rising publication rate. Data programming is a paradigm that circumvents this arduous manual process by combining databases with simple rules and heuristics written as label functions, which are programs designed to annotate textual data automatically. Unfortunately, writing a useful label function requires substantial error analysis and is a nontrivial task that takes multiple days per function. This bottleneck makes populating a knowledge graph with multiple nodes and edge types practically infeasible. Thus, we sought to accelerate the label function creation process by evaluating how label functions can be re-used across multiple edge types.</p><p><strong>Results: </strong>We obtained entity-tagged abstracts and subsetted these entities to only contain compounds, genes, and disease mentions. We extracted sentences containing co-mentions of certain biomedical entities contained in a previously described knowledge graph, Hetionet v1. We trained a baseline model that used database-only label functions and then used a sampling approach to measure how well adding edge-specific or edge-mismatch label function combinations improved over our baseline. Next, we trained a discriminator model to detect sentences that indicated a biomedical relationship and then estimated the number of edge types that could be recalled and added to Hetionet v1. We found that adding edge-mismatch label functions rarely improved relationship extraction, while control edge-specific label functions did. There were two exceptions to this trend, Compound-binds-Gene and Gene-interacts-Gene, which both indicated physical relationships and showed signs of transferability. Across the scenarios tested, discriminative model performance strongly depends on generated annotations. Using the best discriminative model for each edge type, we recalled close to 30% of established edges within Hetionet v1.</p><p><strong>Conclusions: </strong>Our results show that this framework can incorporate novel edges into our source knowledge graph. However, results with label function transfer were mixed. Only label functions describing very similar edge types supported improved performance when transferred. We expect that the continued development of this strategy may provide essential building blocks to populating biomedical knowledge graphs with discoveries, ensuring that these resources include cutting-edge results.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"15 1","pages":"26"},"PeriodicalIF":6.1000,"publicationDate":"2022-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9578183/pdf/","citationCount":"8","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-022-00311-z","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 8
Abstract
Background: Knowledge graphs support biomedical research efforts by providing contextual information for biomedical entities, constructing networks, and supporting the interpretation of high-throughput analyses. These databases are populated via manual curation, which is challenging to scale with an exponentially rising publication rate. Data programming is a paradigm that circumvents this arduous manual process by combining databases with simple rules and heuristics written as label functions, which are programs designed to annotate textual data automatically. Unfortunately, writing a useful label function requires substantial error analysis and is a nontrivial task that takes multiple days per function. This bottleneck makes populating a knowledge graph with multiple nodes and edge types practically infeasible. Thus, we sought to accelerate the label function creation process by evaluating how label functions can be re-used across multiple edge types.
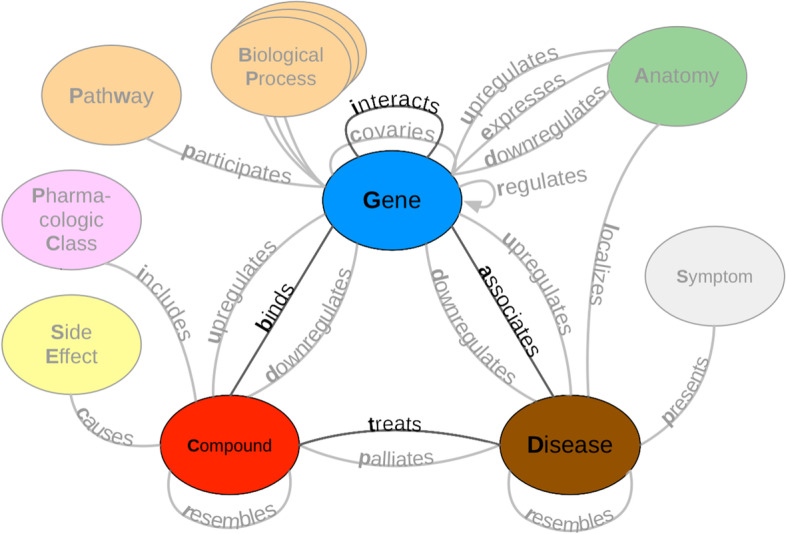
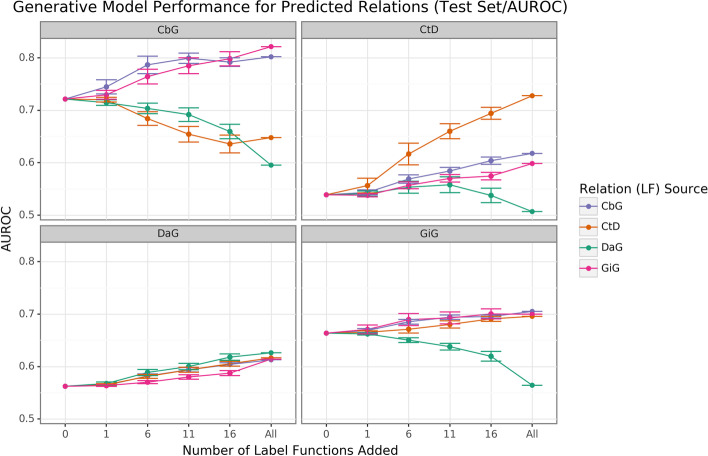
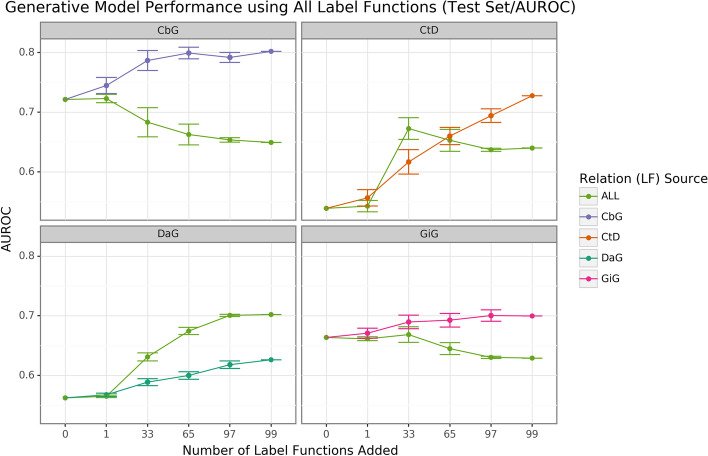
Results: We obtained entity-tagged abstracts and subsetted these entities to only contain compounds, genes, and disease mentions. We extracted sentences containing co-mentions of certain biomedical entities contained in a previously described knowledge graph, Hetionet v1. We trained a baseline model that used database-only label functions and then used a sampling approach to measure how well adding edge-specific or edge-mismatch label function combinations improved over our baseline. Next, we trained a discriminator model to detect sentences that indicated a biomedical relationship and then estimated the number of edge types that could be recalled and added to Hetionet v1. We found that adding edge-mismatch label functions rarely improved relationship extraction, while control edge-specific label functions did. There were two exceptions to this trend, Compound-binds-Gene and Gene-interacts-Gene, which both indicated physical relationships and showed signs of transferability. Across the scenarios tested, discriminative model performance strongly depends on generated annotations. Using the best discriminative model for each edge type, we recalled close to 30% of established edges within Hetionet v1.
Conclusions: Our results show that this framework can incorporate novel edges into our source knowledge graph. However, results with label function transfer were mixed. Only label functions describing very similar edge types supported improved performance when transferred. We expect that the continued development of this strategy may provide essential building blocks to populating biomedical knowledge graphs with discoveries, ensuring that these resources include cutting-edge results.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
