Aljouhra AlHargan, Mohammed A AlMuhaizea, Rawan Almass, Ali H Alwadei, Maha Daghestani, Stefan T Arold, Namik Kaya
{"title":"SHQ1-associated neurodevelopmental disorder: Report of the first homozygous variant in unrelated patients and review of the literature.","authors":"Aljouhra AlHargan, Mohammed A AlMuhaizea, Rawan Almass, Ali H Alwadei, Maha Daghestani, Stefan T Arold, Namik Kaya","doi":"10.1038/s41439-023-00234-z","DOIUrl":null,"url":null,"abstract":"<p><p>Compound heterozygous mutations in SHQ1 have been associated with a rare and severe neurological disorder characterized by global developmental delay (GDD), cerebellar degeneration coupled with seizures, and early-onset dystonia. Currently, only five affected individuals have been documented in the literature. Here, we report three children from two unrelated families harboring a homozygous variant in the gene but with a milder phenotype than previously described. The patients had GDD and seizures. Magnetic resonance imaging analyses revealed diffuse white matter hypomyelination. Sanger sequencing confirmed the whole-exome sequencing results and revealed full segregation of the missense variant (SHQ1:c.833 T > C; p.I278T) in both families. We performed a comprehensive in silico analysis using different prediction classifiers and structural modeling of the variant. Our findings demonstrate that this novel homozygous variant in SHQ1 is likely to be pathogenic and leads to the clinical features observed in our patients.</p>","PeriodicalId":36861,"journal":{"name":"Human Genome Variation","volume":"10 1","pages":"7"},"PeriodicalIF":1.0000,"publicationDate":"2023-02-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9944922/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genome Variation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1038/s41439-023-00234-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
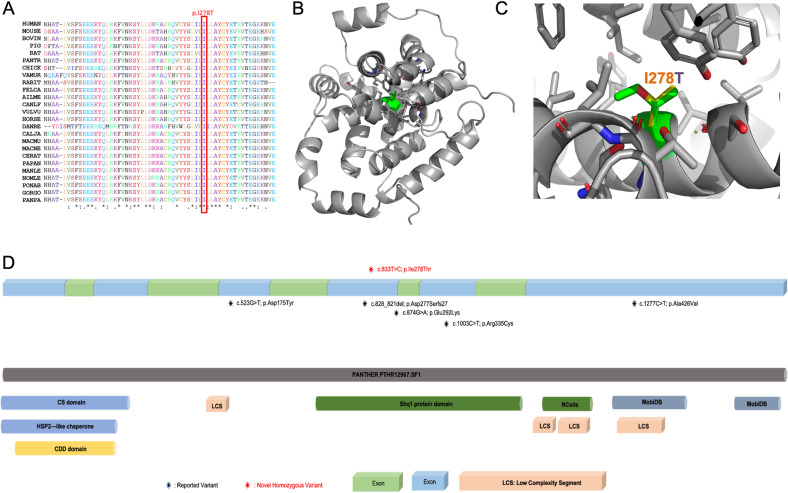
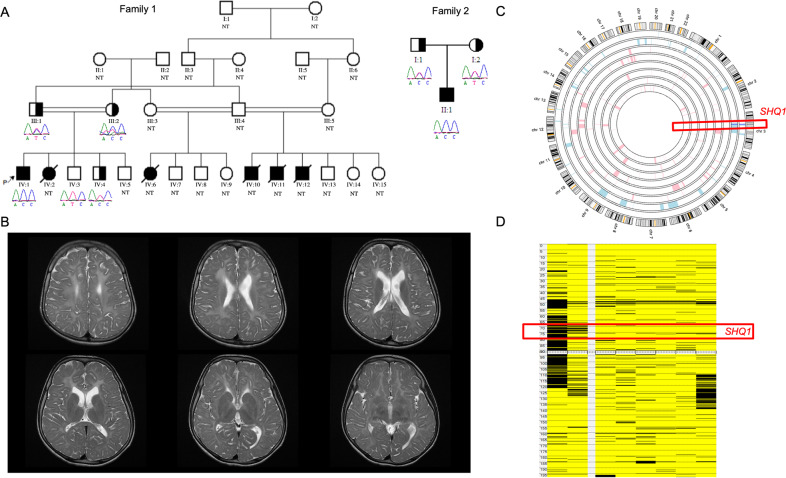
Compound heterozygous mutations in SHQ1 have been associated with a rare and severe neurological disorder characterized by global developmental delay (GDD), cerebellar degeneration coupled with seizures, and early-onset dystonia. Currently, only five affected individuals have been documented in the literature. Here, we report three children from two unrelated families harboring a homozygous variant in the gene but with a milder phenotype than previously described. The patients had GDD and seizures. Magnetic resonance imaging analyses revealed diffuse white matter hypomyelination. Sanger sequencing confirmed the whole-exome sequencing results and revealed full segregation of the missense variant (SHQ1:c.833 T > C; p.I278T) in both families. We performed a comprehensive in silico analysis using different prediction classifiers and structural modeling of the variant. Our findings demonstrate that this novel homozygous variant in SHQ1 is likely to be pathogenic and leads to the clinical features observed in our patients.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
