Ana Solis, Joshua Shimony, Marwan Shinawi, Kevin T Barton
{"title":"Case report: malignant hypertension associated with catecholamine excess in a patient with Leigh syndrome.","authors":"Ana Solis, Joshua Shimony, Marwan Shinawi, Kevin T Barton","doi":"10.1186/s40885-022-00231-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Leigh syndrome is a progressive neurodegenerative mitochondrial disorder caused by multiple genetic etiologies with multisystemic involvement that mostly affecting the central nervous system with high rate of premature mortality.</p><p><strong>Case presentation: </strong>We present a 3-year, 10 month-old female patient with Leigh syndrome complicated by renal tubular acidosis, hypertension, gross motor delay, who presented with hypertensive emergency, persistent tachycardia, insomnia and irritability. Her previous genetic workup revealed a pathogenic variant in the MT-ND5 gene designated as m.13513G > A;p.Asp393Asn with a heteroplasmy of 69%. She presented acutely with malignant hypertension requiring intensive care unit admission. Her acute evaluation revealed elevated serum and urine catecholamines, without an identifiable catecholamine-secreting tumor. After extensive evaluation for secondary causes, she was ultimately found to have progression of her disease with new infarctions in her medulla, pons, and basal ganglia as the most likely etiology of her hypertension. She was discharged home with clonidine, amlodipine and atenolol for hypertension management. This report highlights the need to recognize possible autonomic dysfunction in mitochondrial disease and illustrates the challenges for accurate and prompt diagnosis and subsequent management of the associated manifestations. This association between catecholamine induced autonomic dysfunction and Leigh syndrome has been previously reported only once with MT-ND5 mutation.</p><p><strong>Conclusions: </strong>Elevated catecholamines with malignant secondary hypertension may be unique to this specific mutation or may be a previously unrecognized feature of Leigh syndrome and other mitochondrial complex I deficient syndromes. As such, patients with Leigh syndrome who present with malignant hypertension should be treated without the need for extensive work-up for catecholamine-secreting tumors.</p>","PeriodicalId":10480,"journal":{"name":"Clinical Hypertension","volume":"29 1","pages":"7"},"PeriodicalIF":3.6000,"publicationDate":"2023-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9976414/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Hypertension","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40885-022-00231-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PERIPHERAL VASCULAR DISEASE","Score":null,"Total":0}
引用次数: 1
Abstract
Background: Leigh syndrome is a progressive neurodegenerative mitochondrial disorder caused by multiple genetic etiologies with multisystemic involvement that mostly affecting the central nervous system with high rate of premature mortality.
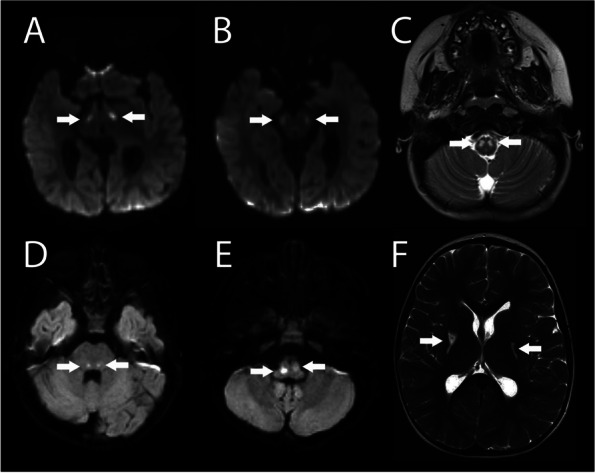
Case presentation: We present a 3-year, 10 month-old female patient with Leigh syndrome complicated by renal tubular acidosis, hypertension, gross motor delay, who presented with hypertensive emergency, persistent tachycardia, insomnia and irritability. Her previous genetic workup revealed a pathogenic variant in the MT-ND5 gene designated as m.13513G > A;p.Asp393Asn with a heteroplasmy of 69%. She presented acutely with malignant hypertension requiring intensive care unit admission. Her acute evaluation revealed elevated serum and urine catecholamines, without an identifiable catecholamine-secreting tumor. After extensive evaluation for secondary causes, she was ultimately found to have progression of her disease with new infarctions in her medulla, pons, and basal ganglia as the most likely etiology of her hypertension. She was discharged home with clonidine, amlodipine and atenolol for hypertension management. This report highlights the need to recognize possible autonomic dysfunction in mitochondrial disease and illustrates the challenges for accurate and prompt diagnosis and subsequent management of the associated manifestations. This association between catecholamine induced autonomic dysfunction and Leigh syndrome has been previously reported only once with MT-ND5 mutation.
Conclusions: Elevated catecholamines with malignant secondary hypertension may be unique to this specific mutation or may be a previously unrecognized feature of Leigh syndrome and other mitochondrial complex I deficient syndromes. As such, patients with Leigh syndrome who present with malignant hypertension should be treated without the need for extensive work-up for catecholamine-secreting tumors.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
