{"title":"Exploration of errors in variance caused by using the first-order approximation in Mendelian randomization.","authors":"Hakin Kim, Kunhee Kim, Buhm Han","doi":"10.5808/gi.21060","DOIUrl":null,"url":null,"abstract":"<p><p>Mendelian randomization (MR) uses genetic variation as a natural experiment to investigate the causal effects of modifiable risk factors (exposures) on outcomes. Two-sample Mendelian randomization (2SMR) is widely used to measure causal effects between exposures and outcomes via genome-wide association studies. 2SMR can increase statistical power by utilizing summary statistics from large consortia such as the UK Biobank. However, the first-order term approximation of standard error is commonly used when applying 2SMR. This approximation can underestimate the variance of causal effects in MR, which can lead to an increased false-positive rate. An alternative is to use the second-order approximation of the standard error, which can considerably correct for the deviation of the first-order approximation. In this study, we simulated MR to show the degree to which the first-order approximation underestimates the variance. We show that depending on the specific situation, the first-order approximation can underestimate the variance almost by half when compared to the true variance, whereas the second-order approximation is robust and accurate.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":"20 1","pages":"e9"},"PeriodicalIF":0.0000,"publicationDate":"2022-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9002003/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.21060","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/3/31 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
Abstract
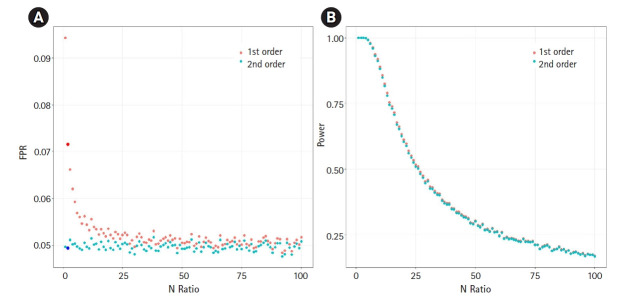
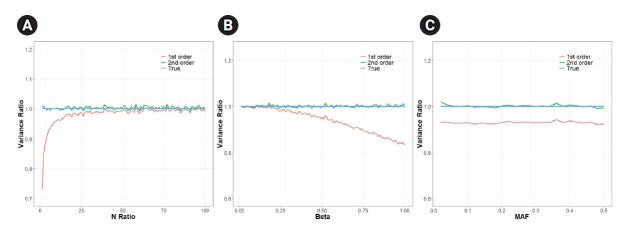
Mendelian randomization (MR) uses genetic variation as a natural experiment to investigate the causal effects of modifiable risk factors (exposures) on outcomes. Two-sample Mendelian randomization (2SMR) is widely used to measure causal effects between exposures and outcomes via genome-wide association studies. 2SMR can increase statistical power by utilizing summary statistics from large consortia such as the UK Biobank. However, the first-order term approximation of standard error is commonly used when applying 2SMR. This approximation can underestimate the variance of causal effects in MR, which can lead to an increased false-positive rate. An alternative is to use the second-order approximation of the standard error, which can considerably correct for the deviation of the first-order approximation. In this study, we simulated MR to show the degree to which the first-order approximation underestimates the variance. We show that depending on the specific situation, the first-order approximation can underestimate the variance almost by half when compared to the true variance, whereas the second-order approximation is robust and accurate.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
